Introduction
Mitochondria are the only intracellular structures,
besides the mammalian nucleus, that possess their own genetic
material. Although the mitochondrial genome is small, mitochondrial
DNA (mtDNA) self-replicates, controlling the function of the
organelle under the direction and regulation of proteins encoded by
nuclear genes. Therefore, mtDNA plays a pivotal role in regulating
the function of mitochondria. Due to the lack of protective
histones, DNA-binding proteins and DNA repair systems, mtDNA has a
much higher rate of mutation compared to nuclear DNA (1–3). The
majority of alterations are heteroplasmic, as both mutant and
wild-type mtDNA coexist within the cells. Mitochondrial dysfunction
caused by mtDNA mutations, such as point mutations, insertions and
deletions, has been implicated in aging (4,5),
mitochondrial respiratory chain disorders such as Kearns-Sayre
syndrome (KSS), progressive external ophthalmoplegia, adult-onset
diabetes mellitus with deafness, Pearson syndrome (6–9),
neurodegenerative diseases such as Alzheimer’s disease (AD),
Parkinson’s disease and Huntington’s disease (10), liver cirrhosis (11,12),
aggressiveness and tumor progression (13–15).
Biliary obstruction, also known as obstructive
jaundice (OJ) and cholestasis, is known to induce numerous
pathophysiological changes (16).
Bile flow may be impaired anywhere between the liver cell
canaliculus and the ampulla of Vater, and may be caused by
intrahepatic or extrahepatic factors, such as stones and tumors.
Only a few reports are currently available on the mechanism by
which OJ affects the morphology and function of mitochondria
(17,18). In addition, little is known
regarding the correlation between OJ and changes in mtDNA in
hepatocytes. Goncalves et al(19) found selective defects in complexes
I, III and IV, which were encoded by mtDNA in two siblings who had
presented with neonatal cholestasis and early liver insufficiency.
Certain investigators (12,20)
have found that the mtDNA copy number decreased following bile duct
ligation (BDL). We hypothesized that damage to mtDNA results, not
only in changes to mtDNA copy number, but also in structural
differences that then affect the normal function of
mitochondria.
In the present study, we investigated the changes in
mtDNA structure and copy number following BDL in rats, and the
effects of such changes on mitochondrial and hepatic function. The
findings may be helpful in understanding the mechanism of altered
hepatic function in OJ.
Materials and methods
Animals and experimental groups
Forty-eight healthy male Wistar rats, provided by
the Animal Center of The Third Military Medical University,
weighing 175–330 g, were used for the experiments. All animals were
housed in a temperature-controlled room on a normal 12-h light/dark
cycle and received a normal diet ad libitum. The animals
were fasted for 12 h prior to sacrifice or surgery.
During the surgery, rats were anesthetized lightly
with ether. The abdominal skin was washed with soap and sterile
water, and then disinfected with iodine tincture. The abdomen was
opened through a midline incision. Forty-eight rats were randomized
into two groups: i) for BDL, the common bile duct was exteriorized
and double ligated close to the liver, and excised just below the
confluence of the lobular ducts; ii) for the Sham operation (Sham),
the common bile duct was exteriorized, and then the abdomen was
closed. Following recovery from anesthesia, each rat was housed in
individual cages and received a normal diet ad libitum. Rats
were sacrificed by decapitation on days 1, 4, 7 and 14 following
surgery. At each time-point, six live rats were used. Blood samples
(3–5 ml) were obtained from the inferior vena cava, and livers were
excised and stored in liquid nitrogen at the designated times. The
serum was separated by centrifugation and stored at −70°C until
use. All animal procedures were approved by the Animal Care and Use
Committee of the Third Military Medical University and conformed to
the Guide for the Care and Use of Laboratory Animals obtained from
the National Institutes of Health. We ensured that animals did not
suffer unnecessarily at any stage of the experiments, whether
acutely or chronically.
Analysis of changes in mitochondrial
function
To determine the effect of damaged mtDNA on
mitochondrial function, a Clark oxygen electrode was used to
measure changes in the mitochondrial respiratory control rate (RCR)
and the phosphorus/oxygen (P/O) value (21). In addition, the ATP content in the
liver was determined using the method of Kimmich et
al(22).
Analysis of hepatic function
Following surgery, blood samples were collected from
the two groups at each time-point and levels of alanine
aminotransferase (ALT), aspartate aminotransferase (AST), serum
total bilirubin (TBIL) and albumin (Alb) were measured to assess
hepatic function.
Extraction of total liver DNA
Total DNA was extracted from samples (30 mg liver
tissue) from all rats in each group according to standard
protocols. DNA extraction kits were purchased from Tianwei Biotech
Co., Ltd. (Beijing, China). In addition, a search for deletions was
performed on rats, according to the manufacturer’s
instructions.
Localization of mtDNA deletions by
amplification of the full-length mitochondrial genome
The entire mitochondrial genome was amplified
according to the long and accurate PCR (LA-PCR) method (23). Primers were designed and
synthesized according to the full-length sequence of mtDNA (gi:
5835177). The following oligonucleotides were used: the forward
primer P1 (nucleotide positions 709–736) was 5′-AGG CAC TAA AGT AAG
CAC AAG AAC AAA C-3′, and the reverse primer P2 (nucleotide
positions 16256–16283) was 5′-TTT CTG AGG GTA GGC AGG TAA AGA GGG
T-3′. The product length was 15,575 bp.
A biphasic hot-start amplification was employed
using 50 μl PCR gems (Takara LA PCRTM kit). The bottom phase of the
PCR mixture (30 μl) contained 23 μl of H2O, 6 μl of
dNTPs (300 μM) and 0.5 μl of P1 and P2 primers (final
concentrations were 0.2 μM). A PCR gem™ (the top phase) was melted
at 80°C for 5 min and placed on top of the bottom phase. The top
phase consisted of 13.5 μl of H2O, 5 μl of 10× LA PCR™
buffer (Mg2+, 2.5 μM), 1 μl of genomic DNA as the target
(150 ng) and 0.5 μl of LA Taq™ (2.5 units). The PCR profile
consisted of an initial 2 min denaturation at 94°C, followed by
two-step PCR for 30 cycles using 20-sec denaturation at 94°C, 15
min annealing/extension at 68°C and a final extension of 10 min at
72°C in a PE2700 thermal cycler (Perkin-Elmer, Waltham, MA, USA).
PCR products (5 μl) were directly loaded onto 0.8% agarose gels
containing ethidium bromide. Following electrophoresis, bands were
visualized by UV light, and the results were recorded with a video
camera.
Restriction enzyme digestion
PCR product (10 μl) was digested with restriction
endonucleases SacI, ApaI (Takara Bio, Kyoto, Japan)
and SauI (Dingguo Biotech Co., Ltd., China) for 90 min at
37°C, electrophoresed through 0.8% agarose gels for 60 min at 4
V/cm and analyzed under a UV light.
Localization of mtDNA deletions
Primers were designed and synthesized according to
the primary location of mtDNA deletions and the full-length
sequence of mtDNA (GI: 5835177). The following oligonucleotides
were used: the upstream primer P3 (nucleotide positions 3878–3900)
was 5′-CCC TTC CCG TAC TAA TAA ATC CA-3′, and the downstream primer
P4 (nucleotide positions 15668–15690) was 5′-GTT GAT TTC ACG GAG
GAT GGT AG-3′. The PCR reaction mixture (50 μl) consisted of 38.5
μl of H2O, 6 μl of dNTP (300 μM), 0.5 μl of each primer
(0.2 μM), 5 μl of 10× LA PCR buffer (Mg2+, 2.5 μM), 1 μl
of genomic DNA as the target (150 ng) and 0.5 μl of LA Taq (2.5
units). The PCR profile consisted of an initial 2-min denaturation
at 94°C, followed by PCR for 40 cycles using 20-sec denaturation at
94°C, 30 sec annealing at 55°C, 1-min extension at 72°C and a final
extension of 10 min at 72°C using the GeneAmp PCR system 2700
(Applied Biosystems, Foster City, CA, USA). Under these conditions,
the extension time was too short to synthesize a product of
>11.8 kb in length, and no product was generated. If a 1.1-kb
mtDNA deletion existed, a DNA fragment of 600–700 bp in length
would have been obtained. Following the PCR reaction, the products
(10 μl) were electrophoresed through 1.0% agarose gels for 30 min
at 5 V/cm and analyzed under UV light.
Quantitation of mtDNA deletions
Real-time quantitative PCR (LightCycler, Roche,
Indianapolis, IN, USA) was used to quantify the relative ratio of
the specific mtDNA deletion and total mtDNA. Primer pairs were
designed, and the following oligonucleotides were used. i) To
quantify the deletions, the deletion-specific PCR product was
amplified using the primer set P3 and P4. ii) For total mtDNA, the
upstream primer P5 (nucleotide positions 2813–2834) was 5′-GGC TAC
ATA CAA TTA CGC AAA G-3′ and the downstream primer P6 (nucleotide
positions 3058–3079) was 5′-TAG AAT GGA GTA GAC CGA AAG G-3′. The
amplified fragment length was 267 bp. The primer pair P5 and P6 was
directed to the ND1 region of the mitochondrial gene, which is a
highly conserved sequence. As a result, the PCR product reflects
the amount of total mtDNA. iii) For β-actin, the upstream primer P7
(nucleotide positions 2747–2771) was 5′-ATC CGT AAA GAC CTC TAT GCC
AAC A-3′, and the downstream primer P8 (nucleotide positions
2900–2923) was 5′-GGC TAC AAC TAC AGG GCT GAC CAC-3′. The amplified
fragment length was 177 bp. Each PCR was performed using the SYBR
Premix Ex Taq™ reagent (Takara Bio), according to the
manufacturer’s instructions (Table
I). The PCR mixture (20 μl) consisted of 10 μl of SYBR Premix
Ex Taq, 2 μl of mtDNA as the target (100 ng), 0.4 μl of each primer
(final concentrations were 0.2 μM) and 7.2 μl of
H2O.
 | Table IConditions of fluorescent quantitative
PCR conditions of mtDNA in hepatocytes of rats with OJ. |
Table I
Conditions of fluorescent quantitative
PCR conditions of mtDNA in hepatocytes of rats with OJ.
| Fluorescent
quantitative PCR conditions |
|---|
|
|
|---|
| Stage 1:
force-degeneration (1 cycle) | Stage 2: PCR (45
cycles) | Stage 3: dilapsus
curve |
|---|
|
|
|
|
|---|
| Product | TTp | TTm | TTR | TTp | TTm | TTR | TTp | TTm | TTR |
|---|
| Total mtDNA
(ND1) | 95 | 60 | 20 | 95 | 10 | 20 | 95 | 0 | 20 |
| | | | 55 | 20 | 20 | 65 | 15 | 20 |
| | | | 72 | 15 | 20 | 95 | 0 | 0.1 |
| Deletion from
mtDNA | 95 | 60 | 20 | 95 | 10 | 20 | 95 | 0 | 20 |
| | | | 55 | 30 | 20 | 65 | 15 | 20 |
| | | | 72 | 60 | 20 | 95 | 0 | 0.1 |
| β-actin | 95 | 60 | 20 | 95 | 10 | 20 | 95 | 0 | 20 |
| | | | 55 | 20 | 20 | 65 | 15 | 20 |
| | | | 72 | 15 | 20 | 95 | 0 | 0.1 |
Standard curves for mtDNA (ND1) and β-actin were
generated with normal control rats. Another standard curve for
deletion-specific mtDNA was generated from rats 14 days after bile
duct obstruction, when liver function was the most severely
impaired. Templates in each reaction were diluted 1:100,000,
1:10,000, 1:1,000, 1:100, 1:10 and 1:1. Substrate concentrations
were calculated according to the standard curves. The ratio of
deletion-specific mtDNA to total mtDNA was determined as the
relative amount of deletion-specific mtDNA in hepatocytes. The
ratio of total mtDNA to β-actin was considered to be the relative
amount of mtDNA copies in liver cells.
Statistical analysis
Variances within the data were determined with the
F-test for variance. Based on these results, the Student’s t-test
for equal or unequal variance was used to determine the statistical
significance between different groups. All tests were two-sided and
P<0.05 was considered to be statistically significant.
Statistical software SPSS12.0 was used.
Results
Analysis of mitochondrial function
The mitochondrial function of the BDL group was
badly compromised as is observed by the lower RCR, P/O ratio and
ATP content in hepatic tissue, compared to the Sham group
(P<0.01) at each time-point. This damage became more pronounced
as the ligation time increased (Table
II).
| Table IIChanges in hepatocyte mitochondrial
function in rats with OJ (mean ± SD). |
Table II
Changes in hepatocyte mitochondrial
function in rats with OJ (mean ± SD).
| Group | Time (days) | RCR | P/O | ATP (μmol/g) |
|---|
| Sham | 1 | 4.03±0.09 | 1.73±0.06 | 2.99±0.10 |
| 4 | 4.03±0.09 | 1.73±0.05 | 3.05±0.06 |
| 7 | 4.05±0.11 | 1.75±0.05 | 3.02±0.10 |
| 14 | 4.02±0.11 | 1.74±0.04 | 3.03±0.11 |
| BDL | 1 | 3.30±0.15a,b | 1.43±0.04a,b | 2.21±0.13a,b |
| 4 | 3.06±0.11a,b | 1.36±0.04a,b | 1.88±0.05a,b |
| 7 | 2.70±0.17a,b | 1.23±0.03a,b | 1.34±0.10a,b |
| 14 | 2.13±0.14a,b | 0.98±0.05a,b | 0.46±0.07a,b |
Changes in hepatic function
In the Sham group, there were no marked changes in
indices of hepatic function, including ALT, AST, Alb and TBIL.
However, in the BDL group, the hepatic function was clearly
impaired, as observed by increases in ALT, AST and TBIL values, and
the decreases in Alb levels, compared to the Sham (P<0.05) at
each time-point. With prolonged ligation time, this damage became
gradually more pronounced (Table
III).
| Table IIIHepatic function parameters in the
two groups (mean ± SD). |
Table III
Hepatic function parameters in the
two groups (mean ± SD).
| Group | Time (days) | AST (IU/l) | ALT (IU/l) | Alb (g/l) | TBIL (μM) |
|---|
| Sham | 1 | 150.00±31.18 | 48.83±4.88 | 34.95±2.36 | 3.68±1.02 |
| 4 | 133.22±12.71 | 45.03±4.54 | 34.72±2.22 | 1.87±1.07 |
| 7 | 167.33±17.03 | 43.33±4.18 | 34.03±2.26 | 4.47±2.19 |
| 14 | 156.83±53.01 | 46.67±4.89 | 35.75±3.83 | 2.51±1.42 |
| BDL | 1 |
1991.83±178.64a | 1176±218.53a | 35.55±1.52 | 69.43±8.80a |
| 4 |
442.67±109.86a | 201.6±33.97a | 28.30±2.22a |
105.30±21.40a |
| 7 |
533.23±75.47a | 279.9±79.04a | 21.35±3.63a |
172.38±36.45a |
| 14 |
824.00±250.21a | 671.±288.29a | 16.50±1.54a |
368.83±52.08a |
Location of mtDNA deletions in
hepatocytes of rats with OJ
In the Sham group, products of ~15.5 kb in length
were found, which are considered to be full-length mtDNA. However,
in the BDL group, besides the full-length product, additional lower
molecular weight products of ~4.4 kb in length were also
identified. This appears to be due to an mtDNA deletion that, to
the best of our knowledge, has not been reported previously
(Fig. 1). Restriction enzyme
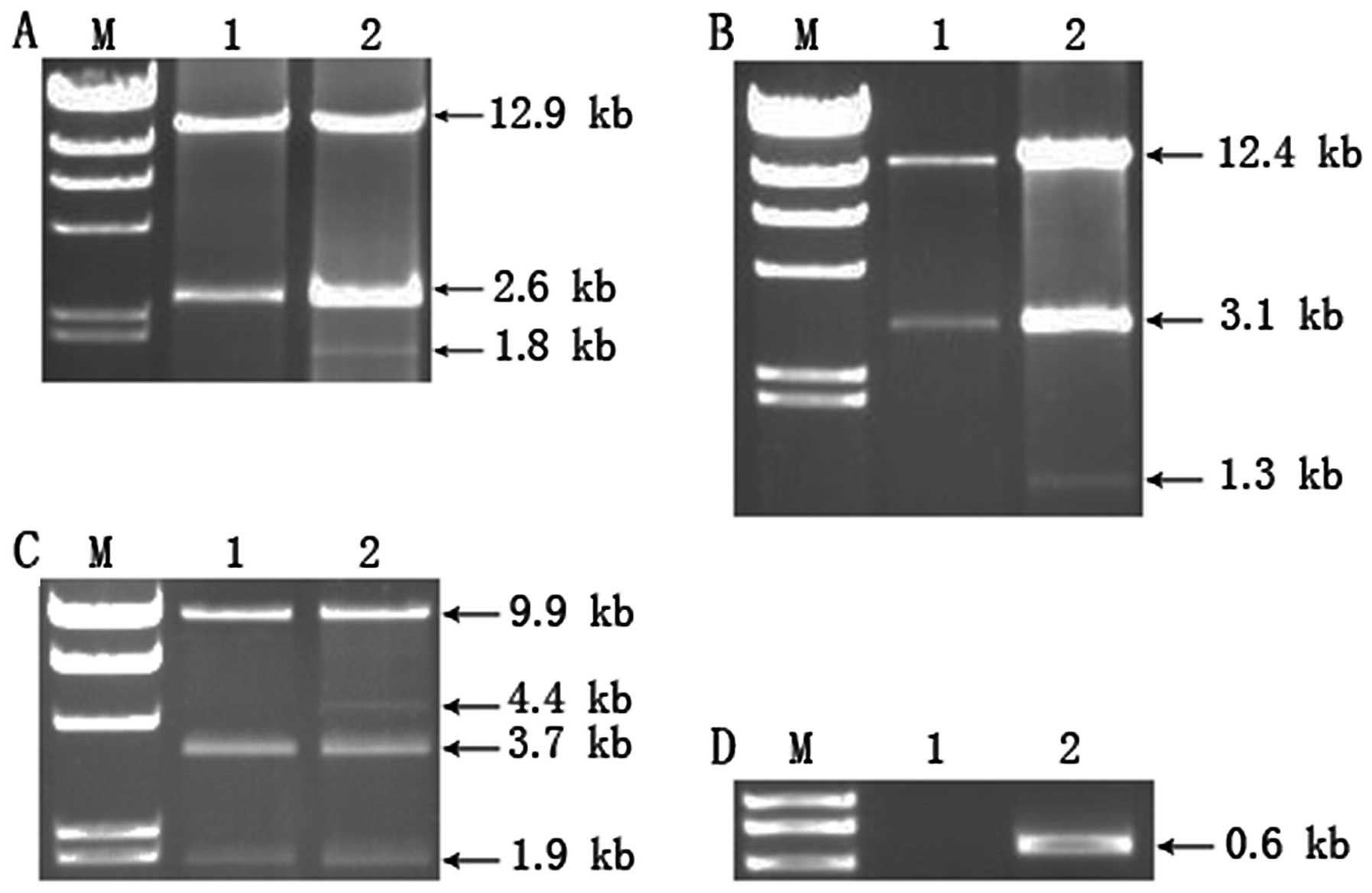
digestion with SacI resulted in 2.6- and 12.9-kb fragments
in the Sham group. However, besides these fragments, an additional
fragment of 1.8 kb was detected in the BDL group (Fig. 2A). This result was consistent with
restriction enzyme mapping of SacI, meaning these PCR
products were full-length or deleted mtDNA. All rats in the BDL
group had this mutation at each time-point.
To determine the location of the mtDNA deletion, PCR
products were digested with restriction endonucleases ApaI
and SauI. Digestion with ApaI resulted in 3.1- and
12.4-kb fragments in the Sham group, whereas in the BDL group,
besides these two fragments, a 1.3-kb fragment was found (Fig. 2B). SauI digestion showed
that in the Sham group, 1.9-, 3.7- and 9.9-kb fragments were
generated. However, in the BDL group, an additional 4.4-kb fragment
was identified (Fig. 2C).
Comparing the results of SacI and SauI restriction
enzyme digestion, the 4.4-kb fragment was not digested by
SauI, indicating the absence of the restriction site at
nucleotide position 4390 in the deleted mtDNA. Thus, the start of
the mtDNA deletion is located between nucleotide positions 3854 and
4390 and the end site is located between nucleotide positions 14954
and 15490.
To determine the accurate location of the mtDNA
deletion, primers were designed according to the primary location
of the specific mtDNA deletion, and the PCR was performed with a
10-min extension time, which did not result in the generation of
the full-length mtDNA. Following PCR amplification, a 600–700-bp
fragment was generated in the BDL group, whereas no fragment was
generated in the Sham group (Fig.
2D). The amplified PCR product was sent to Biotech Company
(Shanghai, China) for sequencing by GeneRay. The result showed that
the sequence of the fragment was exactly the same as that of mtDNA
at nucleotide positions 3878–4100 and nucleotide positions
15295–15690. These results suggested the presence of a large
(11,194 bp) mtDNA deletion in the BDL group, which was located at
nucleotide positions 4101–15294.
Quantification of the mtDNA deletion in
hepatocytes
In all rats in the BDL group the copy number of the
total mtDNA was significantly lower (P<0.01), compared to the
Sham group at the same time-point (Table IV). With prolonged ligation time,
the decrease became more pronounced, and the ratio of
deletion-specific mtDNA to total mtDNA in the BDL group increased.
The difference was also statistically significant (P<0.01). In
the Sham group, no mtDNA deletion was found.
| Table IVQuantitative changes of mtDNA in
hepatocytes of rats with OJ (mean ± SD). |
Table IV
Quantitative changes of mtDNA in
hepatocytes of rats with OJ (mean ± SD).
| Group | Time (days) | Total mtDNA
(ND1) | Deleted mtDNA
(%) |
|---|
| Sham | 1 | 4.39±0.54 | - |
| 4 | 4.67±0.44 | - |
| 7 | 4.69±0.73 | - |
| 14 | 4.31±0.51 | - |
| BDL | 1 | 2.50±0.61a,b | 4.44±1.47b |
| 4 | 2.15±0.33a,b | 11.19±3.31b |
| 7 | 1.64±0.30a,b | 19.64±3.03b |
| 14 | 1.16±0.33a,b | 30.24±3.85b |
Discussion
mtDNA is a double-stranded DNA ring that is packed
into a nucleo-protein complex, known as a nucleoid (24,25).
mtDNA lacks introns and encodes 13 proteins necessary for assembly
of the respiratory chain, 22 tRNAs and two ribosomal RNAs (26). Due to inefficiencies in the DNA
repair system and the intrinsic characteristics of mtDNA, it
represents a critical cellular target for damage and is more
susceptible to damage, compared to genomic DNA. The main reasons
for this observation are: i) due to the lack of protective
histones, mtDNA is almost ‘naked’ and is easily affected by
external factors. ii) The ratio of lipid to mtDNA is high, leading
to preferential accumulation of lipophilic carcinogens on mtDNA.
iii) mtDNA is always replicating during the cell cycle,
contributing to its poor stability. iv) The stability of mtDNA is
constantly challenged by the endogenous production of mitochondrial
reactive oxygen species (mtROS), which are generated during normal
electron flux through mitochondrial electron transport. Unlike
nuclear DNA, mtDNA is located in proximity to mtROS generation
sites, which are located within complexes I and III of the electron
transport chain (27). In fact,
the steady-state levels of oxidatively induced lesions observed in
mtDNA may be several-fold higher than those in nuclear DNA
(28). Due to the mutagenic nature
of many of the ROS-induced lesions, mitochondrial-free radicals are
thought to be an important source of mtDNA mutations and DNA
instability (5). The proofreading
activity of DNA polymerase γ and the formation of a hairpin
structure in the region of the tRNA gene cause high rates of
mispairing during mtDNA replication (29).
Due to the characteristics mentioned above and the
significance of mtDNA in regulating mitochondrial function, various
studies (30) have shown that
alterations in mtDNA, such as point mutations, insertion mutations
and deletions, are associated with aging and many diseases in
post-mitotic tissues, including skeletal muscle, heart and brain,
all of which are heavily dependent on intact functional
mitochondria. However, few studies have been conducted to provide
insight into the effect of OJ on mtDNA, particularly on the
structure of mtDNA.
We hypothesized that following BDL, the combination
of increased ROS generation and impaired antioxidant capacity
resulted in mtDNA damage. mtDNA damage may involve various
pathways, leading to the generation of a large deletion.
Typically, a single cell contains hundreds of
mitochondria with multiple copies (2–10) of
mtDNA, which are required to maintain normal respiratory function
in mitochondria (31). mtDNA
mutation-induced pathological consequences in a particular organ or
tissue often require a minimum critical amount of damaged mtDNA
(32–34). The amount of damaged mtDNA was also
measured by real-time quantitative PCR. The results showed, not
only a mtDNA deletion, but also a decrease in mtDNA copy number in
the BDL group. With increasing ligation time, the damage to mtDNA
became increasingly clear as the ratio of damaged mtDNA to total
mtDNA in the BDL group increased. This mtDNA damage correlated with
worsening hepatic function.
In conclusion, our results have confirmed the
existence of a specific 11,194 bp mtDNA deletion located at
nucleotide positions 4101–15,294 in hepatocytes of rats with OJ and
a decrease of mtDNA copies. Our study gives insight into the
correlation between mtDNA deletion and OJ and provides a
mechanistic understanding of hepatic function affected by OJ. This
study provides information that may be useful during the
perioperative period in protecting and improving impaired hepatic
function caused by OJ.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grants no. 30571806, 30872494 and
30801114).
References
|
1
|
Wallace DC: Mitochondrial DNA sequence
variation in human evolution and disease. Proc Natl Acad Sci USA.
91:8739–8746. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brown WM, George M Jr and Wilson AC: Rapid
evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA.
76:1967–1971. 1979. View Article : Google Scholar
|
|
3
|
Wallace DC: 1994 William Allan Award
Address. Mitochondrial DNA variation in human evolution,
degenerative disease, and aging. Am J Hum Genet. 57:201–223.
1995.PubMed/NCBI
|
|
4
|
Linnane AW, Marzuki S, Ozawa T and Tanaka
M: Mitochondrial DNA mutations as an important contributor to
ageing and degenerative diseases. Lancet. 1:642–645. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hartmann N, Reichwald K, Wittig I, et al:
Mitochondrial DNA copy number and function decrease with age in the
short-lived fish Nothobranchius furzeri. Aging Cell. 10:824–831.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ballinger SW, Shoffner JM, Hedaya EV, et
al: Maternally transmitted diabetes and deafness associated with a
10.4 kb mitochondrial DNA deletion. Nat Genet. 1:11–15. 1992.
View Article : Google Scholar
|
|
7
|
Wallace DC: Diseases of the mitochondrial
DNA. Annu Rev Biochem. 61:1175–1212. 1992. View Article : Google Scholar
|
|
8
|
Superti-Furga A, Schoenle E, Tuchschmid P,
et al: Pearson bone marrow-pancreas syndrome with insulin-dependent
diabetes, progressive renal tubulopathy, organic aciduria and
elevated fetal haemoglobin caused by deletion and duplication of
mitochondrial DNA. Eur J Pediatr. 152:44–50. 1993. View Article : Google Scholar
|
|
9
|
Rotig A, Bourgeron T, Chretien D, Rustin P
and Munnich A: Spectrum of mitochondrial DNA rearrangements in the
Pearson marrow-pancreas syndrome. Hum Mol Genet. 4:1327–1330. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Beal MF: Mitochondria take center stage in
aging and neurodegeneration. Ann Neurol. 58:495–505. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamamoto H, Tanaka M, Katayama M, Obayashi
T, Nimura Y and Ozawa T: Significant existence of deleted
mitochondrial DNA in cirrhotic liver surrounding hepatic tumor.
Biochem Biophys Res Commun. 182:913–920. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arduini A, Serviddio G, Escobar J, et al:
Mitochondrial biogenesis fails in secondary biliary cirrhosis in
rats leading to mitochondrial DNA depletion and deletions. Am J
Physiol Gastrointest Liver Physiol. 301:G119–G127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chinnery PF, Samuels DC, Elson J and
Turnbull DM: Accumulation of mitochondrial DNA mutations in ageing,
cancer, and mitochondrial disease: is there a common mechanism?
Lancet. 360:1323–1325. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Amuthan G, Biswas G, Zhang SY,
Klein-Szanto A, Vijayasarathy C and Avadhani NG:
Mitochondria-to-nucleus stress signaling induces phenotypic
changes, tumor progression and cell invasion. EMBO J. 20:1910–1920.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sotgia F, Martinez-Outschoorn UE and
Lisanti MP: Mitochondrial oxidative stress drives tumor progression
and metastasis: should we use antioxidants as a key component of
cancer treatment and prevention? BMC Med. 9:622011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moritz M and Snodgrass PJ: Serum enzymes
derived from liver cell fractions. II Responses to bile duct
ligation in rats. Gastroenterology. 62:93–100. 1972.PubMed/NCBI
|
|
17
|
Kim YH and Joo II: Arylamine
N-methyltransferase and thiol methyltransferase activities in
cholestatic rat liver induced by common bile duct ligation. Exp Mol
Med. 33:23–28. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang YJ, Iwata S, Terada Y and Ozawa K:
Restricted redox oscillation in oxidative phosphorylation in
jaundiced rat liver mitochondria and its relation to calcium ion. J
Surg Res. 66:91–99. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goncalves I, Hermans D, Chretien D, et al:
Mitochondrial respiratory chain defect: a new etiology for neonatal
cholestasis and early liver insufficiency. J Hepatol. 23:290–294.
1995.PubMed/NCBI
|
|
20
|
Tiao MM, Lin TK, Liou CW, et al: Early
transcriptional deregulation of hepatic mitochondrial biogenesis
and its consequent effects on murine cholestatic liver injury.
Apoptosis. 14:890–899. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Estabrook RW: Mitochondrial respiratory
control and the polarographic measurement of ADP: O ratios. Methods
Enzymol. 10:41–47. 1967. View Article : Google Scholar
|
|
22
|
Kimmich GA, Randles J and Brand JS: Assay
of picomole amounts of ATP, ADP, and AMP using the luciferase
enzyme system. Anal Biochem. 69:187–206. 1975. View Article : Google Scholar
|
|
23
|
Melov S, Shoffner JM, Kaufman A and
Wallace DC: Marked increase in the number and variety of
mitochondrial DNA rearrangements in aging human skeletal muscle.
Nucleic Acids Res. 23:4122–4126. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holt IJ, He J, Mao CC, et al: Mammalian
mitochondrial nucleoids: organizing an independently minded genome.
Mitochondrion. 7:311–321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen XJ and Butow RA: The organization and
inheritance of the mitochondrial genome. Nat Rev Genet. 6:815–825.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Graeber MB, Grasbon-Frodl E, Eitzen UV and
Kosel S: Neurodegeneration and aging: role of the second genome. J
Neurosci Res. 52:1–6. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Muller FL, Liu Y and Van Remmen H: Complex
III releases superoxide to both sides of the inner mitochondrial
membrane. J Biol Chem. 279:49064–49073. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hamilton ML, Guo Z, Fuller CD, et al: A
reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and
mitochondrial DNA using the sodium iodide method to isolate DNA.
Nucleic Acids Res. 29:2117–2126. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pinz KG, Shibutani S and Bogenhagen DF:
Action of mitochondrial DNA polymerase gamma at sites of base loss
or oxidative damage. J Biol Chem. 270:9202–9206. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wallace DC: Mitochondrial DNA mutations in
disease and aging. Environ Mol Mutagen. 51:440–450. 2010.PubMed/NCBI
|
|
31
|
DiMauro S and Moraes CT: Mitochondrial
encephalomyopathies. Arch Neurol. 50:1197–1208. 1993. View Article : Google Scholar
|
|
32
|
DiMauro S: Mitochondrial diseases. Biochim
Biophys Acta. 1658:80–88. 2004. View Article : Google Scholar
|
|
33
|
Porteous WK, James AM, Sheard PW, et al:
Bioenergetic consequences of accumulating the common 4977-bp
mitochondrial DNA deletion. Eur J Biochem. 257:192–201. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Carling PJ, Cree LM and Chinnery PF: The
implications of mitochondrial DNA copy number regulation during
embryogenesis. Mitochondrion. 11:686–692. 2011. View Article : Google Scholar : PubMed/NCBI
|