Introduction
The epigenetic programming of the gametes and early
post-fertilization embryos is essential for the development of a
new organism. This programming involves the intricate interaction
of the three layers of epigenetics: DNA methylation, histone
modification and non-coding RNAs (1). Increasing evidence supports the
importance of DNA methylation in the regulation of new organism
development. 5-Methylcytosine (5mC) in the DNA from mammalian cells
is found to be located almost entirely within CpG dinucleotides
(2,3) and 70–80% of cytosine in CpG dyads is
methylated on both strands in human cells. In general, CpG
methylation is highly prevalent in repetitive sequences and in gene
bodies, but rare at CpG islands within housekeeping promoters
(2). Consistent with the
functional importance of DNA methylation, it is non-random,
well-regulated and tissue-specific (4–6). DNA
methylation studies have attracted increasing interest in the field
of epigenetics which is now a dynamic area of research challenging
and revising traditional paradigms of gene expression and behavior
(7).
Complete hydatidiform mole (CHM) is one of the most
frequent abnormalities occurring during pregnancy (8). Although a number of studies have
indicated that CHM is a maternal-effect autosomal recessive
disorder in which recurrent pregnancy failure with molar
degeneration occurs (9), the
detailed mechanism of CHM genesis is not fully understood. The
occurrence of CHM may have an epigenetic link. Hayward et al
revealed a methylation defect at imprinted gene NLRP7 (NALP7) loci
in tissue from four new familial biparental hydatidiform moles
(9) and the differential
methylation of SGCE/PEG10 was preserved in these four cases.
Furthermore, differential methylation at the H19 locus (an
imprinted gene) was observed in two hydatidiform mole cases
(10). Li et al
investigated the expression and methylation profiles of SOX2 in
hydatidiform moles and choriocarcinoma and demonstrated that
epigenetic mechanisms may be important in the transcription
regulation of SOX2 and contribute to the pathogenesis of
hydatidiform moles (11). It is
becoming clear that distinct epigenetic markers are essential for
the pathogenesis of hydatidiform moles.
In the present study, we developed
methylation-specific genome subtractive hybridization (MS-G-SH), a
novel method to screen for methylated biomarkers for the early
diagnosis of CHM. We found several methylated sequences in the
whole genomes from tissue samples which differed between
hydatidiform moles and villi. The methylation of two candidate
genes, insulin-like growth factor 2 (IGF2) and transforming growth
factor-β (TGF-β), was revealed to be related to the pathogenesis of
CHM.
Materials and methods
Clinical sample collection
The trophoblastic tissues (hydatidiform moles and
villi) were collected from the Shanghai First Maternity and Infant
Hospital (Shanghai, China) between June 2009 and March 2010. Cases
suspected by clinical and ultrasonographic analysis to be
hydatidiform moles were suction evacuated. All human samples were
obtained after obtaining approval from the Ethics Review Board of
the Shanghai First Maternity and Infant Hospital and after
obtaining written informed consent from the subjects. Due to
material limitations, we could only analyze a limited number of
hydatidiform moles and villi (3 of each).
Modified NotI-CODE and SSH procedure
The construction of NotI linking libraries
has been previously described (12). Plasmid DNA was purified using the
Axygen plasmid DNA extra kit (Axygen Biosciences, Union City, CA,
USA). A standard protocol was used to prepare nylon filter replicas
of a grid of NotI linking-specific NotI linking
clones and five random unmapped human NotI linking clones.
For hybridization to the nylon filters, the NotI
representing probes were biotin-labeled by PCR. Sequencing gels
were run on ABI 310 automated sequencers (Applied Biosystems,
Carlsbad, CA, USA) according to the manufacturer’s instructions
(12). Two oligonucleotides,
Not-X: 5′-AAAAGAATGTCAGTGTGTCAC GTATGGACGAATTCGC-3′ and Not-Y:
5′-AAACTT ACAGTGTGTGTCACGTATG GCTGCTTAAGCGCCGG-3′, were used to
create the NotI linker. DNA A (tester) and DNA B (driver) (2
mg) at a concentration of 50 μg/ml were digested with 20 units
BamHI and 20 units BglII (Roche Molecular
Biochemicals, Mannheim, Germany) at 37°C for 5 h. After
heat-inactivating for 20 min at 85°C, 0.4 μg digested DNA was
circularized overnight with T4 DNA ligase (Roche Molecular
Biochemicals) in the appropriate buffer in a 1-ml reaction mixture.
The DNA was then concentrated with ethanol, partially filled in and
digested with 10 units NotI at 37°C for 3 h. After
digestion, NotI was heat-inactivated and the DNAs were
ligated overnight in the presence of a 50 M excess of NotI
linker at room temperature. All further steps were performed as
previously described (12), with
the modification that Not-X primer was used for PCR amplification
and only two cycles were performed. These PCR-amplified tester and
driver amplicons are known as NRs.
Quantitative real-time PCR (qRT-PCR)
analysis of mRNA expression
Total RNA from each tissue was isolated using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The RNA samples were treated with
DNase I (Sigma-Aldrich, St. Louis, MO, USA), quantified and
reverse-transcribed into cDNA using the ReverTra Ace-α first strand
cDNA synthesis kit (Toyobo, Osaka, Japan). qRT-PCR was conducted
using a realplex4 real-time PCR detection system from Eppendorf
Co., Ltd., (Hamburg, Germany), using SYBR-Green real-time PCR
Master mix (Toyobo) as the detection dye. qRT-PCR amplification was
performed over 40 cycles with denaturation at 95°C for 15 sec and
annealing at 58°C for 45 sec. The target cDNA was quantified using
the relative quantification method. A comparative threshold cycle
(Ct) was used to determine gene expression relative to a control
(calibrator) and steady-state mRNA levels are reported as an n-fold
difference relative to the calibrator. For each sample, the Ct
values of the marker genes were normalized using the formula ΔCt =
Ctgene - Ct18SRNA. To determine relative
expression levels, the following formula was used ΔΔCt =
ΔCthydatidiform_moles - ΔCtvilli. The values
used to plot the relative expression levels of the markers were
calculated using the expression 2−ΔΔCt. The mRNA levels
were calibrated based on the levels of 18S rRNA. The cDNA of each
gene was amplified using primers as follows: TGF-β forward:
5′-CCCTGGACACCAACTATTGC-3′; TGF-β reverse:
5′-CTTCCAGCCGAGGTCCTT-3′. IGF2 forward: 5′-GTT CGGTTTGCGACACG-3′;
and IGF2 reverse: 5′-AGAAGC ACCAGCATCGACTT-3′.
Bisulfite conversion of genomic DNA and
methylation-specific PCR (MS-PCR)
The cells were lysed with DNA lysis buffer (0.5%
SDS, 0.1 M EDTA, 10 mM Tris-HCl pH 8.0 and 100 ng/ml Proteinase K,
all from Sigma-Aldrich) and incubated at 55°C for 2 h. The
treatment of genomic DNA and the MS-PCR assay were performed as
previously described (13). The
specific primers for TGF-β and IGF2 were designed as follows: TGF-β
methylated forward: 5′-TTTTGTATAATA GTATTCGCGATCG-3′; TGF-β
methylated reverse: 5′-TAA CCTCCTTAACGTAATAATCGAC-3′; TGF-β
unmethylated forward: 5′-TTTGTATAATAGTATTTGTGATTGG-3′; TGF-β
unmethylated reverse: 5′-TAACCTCCTTAACATAAT AATCAAC-3′. IGF2
methylated forward: 5′-GTGTTTTTT ATTAATTTGGCGTTC-3′; IGF2
methylated reverse: 5′-ACT AAAAAATATCCACCAACTCCG-3′; IGF2
unmethylated forward: 5′-TGGTGTTTTTTATTAATTTGGTGTTT-3′; and IGF2
unmethylated reverse: 5′-ACTAAAAAATATCCACCA ACTCCAC-3′. The PCR
products were separated by agarose gel electrophoresis with 12 g/l
ethidium bromide containing 1X Tris-Acetate EDTA (TAE) buffer and
visualized under UV illumination.
Statistical analysis
Each experiment was performed as least three times
and data are shown as the mean ± SE where applicable and
differences were evaluated using the Student’s t-test. P<0.05
was considered to indicate a statistically significant result.
Results
DNA samples subtracted by NotI and
MS-G-SH treatment
If a particular NotI site is present in a DNA
sample, the self-ligated cyclized DNA molecule will be opened with
NotI and labeled; however, if this NotI site is
deleted or methylated, the normal DNA will not contain the
corresponding DNA sequence (12).
Therefore, following NotI subtraction and MS-G-SH, the
methylated NotI sites in the DNA samples will behave as
deleted NotI sites since they will not be digested and
therefore the procedure will simultaneously detect genes that are
either deleted or methylated (12). In addition, the tester and driver
DNA were digested with BamHI and BglII and
self-ligated at DNA concentrations too low for cyclization.
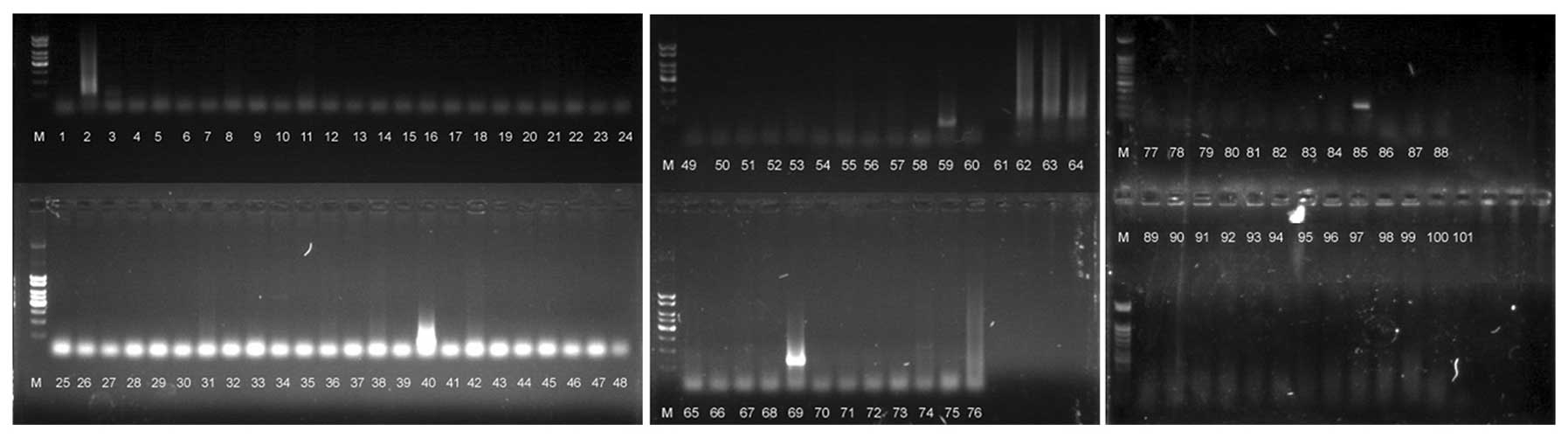
Following NotI subtraction and MS-G-SH treatment, we
isolated the DNA from 101 random clones and sequenced them
(Fig. 1). Eight of these clones
contained the NotI site (numbers 2, 34, 59, 62, 63, 64, 69
and 85). After sequencing, three different clones were determined
to contain NotI sites (C3, A11 and G9). These results
demonstrate the efficiency of subtraction using
NotI-surrounding sequences.
CpG islands were located on the gene
locus
After sequencing, we considered whether the CpG
islands were located on the PCR products. The DNA methylation
status of the three DNA segments was analyzed using CpG island
searcher software (current version: 10/29/04, http://cpgislands.usc.edu/). This analysis revealed
the presence of long CpG islands in the A11 and G9 DNA segments
(Fig. 2). There were ~589 bp CpG
islands (in the 495–1083 bp region) in the A11 DNA product and ~500
bp CpG islands (in the 2–501 bp region) in the G9 DNA product.
However, CpG islands in the C3 DNA segments were not observed. The
findings indicated that two positive CpG island DNA segments which
were modified by DNA methylation would be found by performing
MS-G-SH.
Similarity in products of MS-G-SH and the
human genome were analyzed using the Blast tool
After sequencing and CpG island forecasting, the
Blast tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to
determine which genes of the human genome had high similarity with
the products of MS-G-SSH (Fig. 2).
The analysis results revealed that the nucleic acid sequence of A11
had high similarity to the secondary exon of Homo sapiens
IGF2 (somatomedin A, NM_000612.4). The rate of query coverage was
100% (for the 721–900 bp region of the IGF2 gene) and the percent
identity was 92% (165/180) between the product of MS-G-SSH and the
IGF2 gene. Blast tool analysis also revealed that the G9 sequence
had high homology to the primary exon of Homo sapiens TGF-β1
(NM_000660.4). The rate of query coverage was 95% (for the 102–426
bp region of the TGF-β gene) and the percent identity was 95%
(307/325). According to the results of the Blast tool and CpG
island analysis, the TGF-β and IGF2 genes had specific
modifications of methylation that differed between hydatidiform
moles and villi.
CpG islands of TGF-β and IGF2 were
modified by specific methylation that differed between hydatidiform
moles and villi
Four pairs of primers (two pairs of unmethylated
primers and two pairs of methylated primers) were designed to
determine the methylation status of the primary exon of TGF-β and
the secondary exon of IGF2. Genomic DNA from each group (3
hydatidiform moles and 3 villi) was extracted and positive integral
genomic DNA bands were determined via agarose gel electrophoresis.
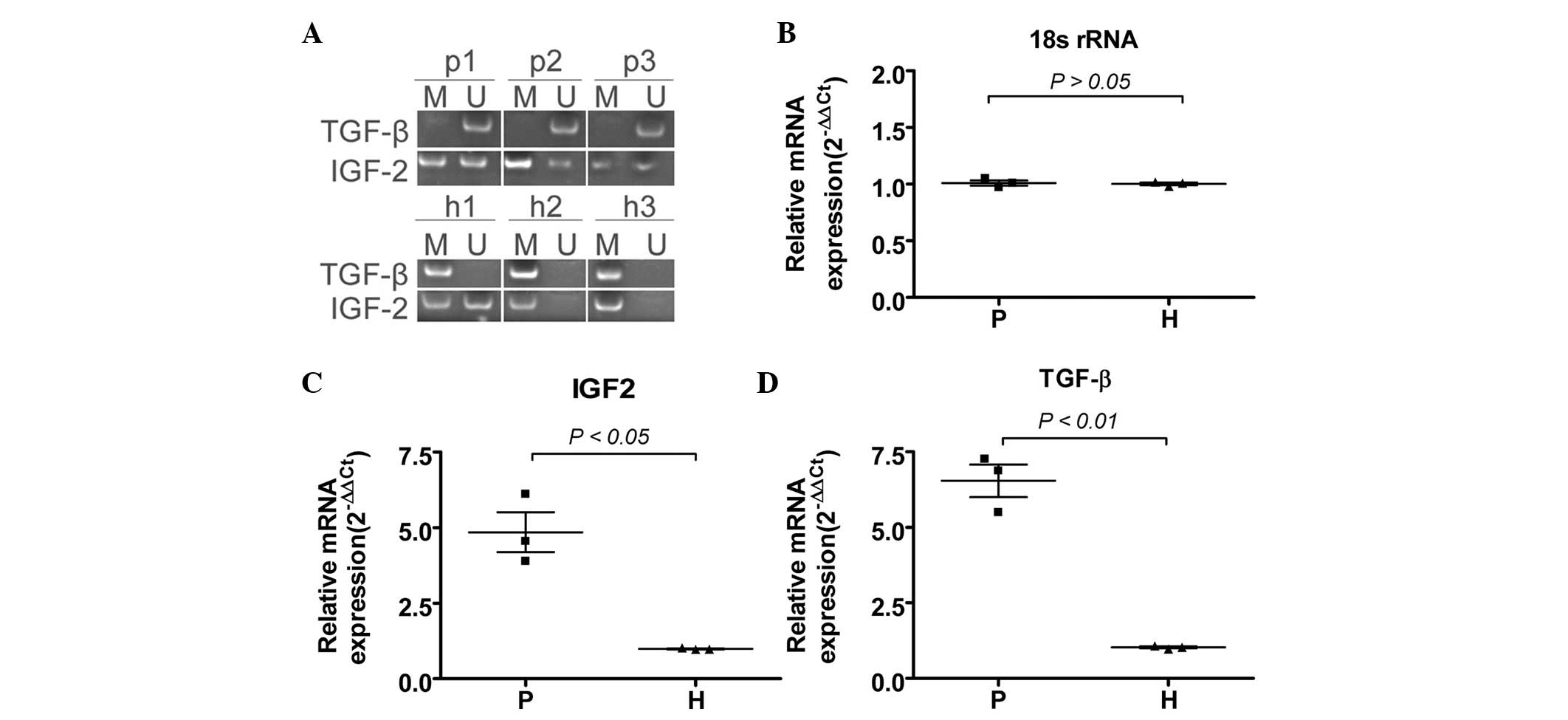
The DNA methylation status of the TGF-β and IGF2 exon CpG islands
was then investigated by sodium bisulfite treatment and sequencing
of the genomic DNA from each group (Fig. 3). In the CpG island region of the
primary exon of TGF-β, there was moderate hypermethylation in three
normal samples of villi but marked hypomethylation in the three
samples of hydatidiform moles. In the CpG island region of the
secondary exon of IGF2, in the normal group, there was marked
hypomethylation in two villi, while the locus of one villus was
partially methylated. However, in the hydatidiform mole group, no
significant changes were observed in the CpG island methylation
status of the specific locus, and all hydatidiform mole samples
displayed partial methylation. These differences between
hydatidiform moles and villi suggest that the exons of TGF-β and
IGF2 were epigenetically modified in a dynamic manner.
mRNAs of TGF-β and IGF2 are expressed
differentially between hydatidiform moles and villi
To determine whether the mRNAs of TGF-β or IGF2
differed when their exon CpG had specific methylated modifications
that differed between the hydatidiform moles or villi, the
expression levels of mRNA were assayed by qRT-PCR. We found that
when the primary exon of TGF-β was moderately hypermethylated in
normal samples of villi, the mRNA expression levels of TGF-β were
significantly lower than those in hydatidiform moles (Fig. 3). Regarding IGF2 expression, we
found that there was marked hypomethylation in three samples of
appreciably higher in hydatidiform moles than in villi. Relative
mRNA expression is shown following normalization to 18S rRNA, which
served as an internal control. These results suggest that when DNA
methylation differed, the mRNA expression level was likely to be
affected.
Discussion
The role of the epigenetic modification of DNA and
histone in the development of human diseases is just beginning to
be understood (14). DNA
methylation studies have attracted increasing interest in the field
of epigenetics, which was poorly understood a few decades ago but
is now a dynamic area of research challenging and revising
traditional paradigms of gene expression and behavior (7). Methods for determining DNA
methylation and histone modification have been developed. To date,
the bisulfite treatment and MS-PCR, and chromatin immunoprecipition
(ChIP) are two promising approaches for determining gene
methylation and histone modification. Bisulfite treatment and
subsequent MS-PCR enables the rapid assessment of the methylation
status of virtually all CpG sites within a CpG island, independent
of the use of methylation-sensitive restriction enzymes (15). In 1996, Herman et al
identified promoter region hypermethylation changes associated with
transcriptional inactivation in four important tumor suppressor
genes (p16, p15, E-cadherin and von Hippel-Lindau) in human cancer
by performing MS-PCR (15). In
addition, ChIP is a powerful experimental approach that enables the
identification of the proteins associated with specific regions of
the genome (16). With the
appropriate antibodies, it may be used to locate non-histone
proteins and histones carrying specific covalent modifications,
including acetylation, phosphorylation or methylation (16). It may also be used to study
molecular mechanisms in transcription, DNA replication, DNA repair
and chromatin remodeling during development and differentiation
(16). However, each technology
used to examine the epigenetic modification of DNA methylation has
its advantages and disadvantages. The largest limitation of MS-PCR
is that it is not suitable for examining DNA methylation in larger
regions. Thus, methylation-sensitive, single-strand conformation
analysis (MS-SSCA) and NotI subtraction and MS-G-SH have
been developed to solve these problems (7,12).
Previous studies have shown that NotI sites are almost
exclusively located in CpG islands and are closely correlated with
functional genes (3,18–23).
Therefore, NotI sites may serve as very useful markers for
physical and genetic mapping and also for the examination of DNA
methylation. In 2002, Li et al used MS-G-SH in combination
with microarrays to detect copy number and methylation changes in
the whole genomes of human cancer cells (12). MS-G-SH was developed to screen a
relatively large number of CpG sites on a CpG island; moreover, it
may be targeted to regions of particular importance within the
island.
In the present study, the MS-G-SH technique has been
improved by performing MS-PCR to examine DNA product methylation
instead of predigestion. MS-G-SH and MS-PCR have been applied to
detect DNA methylation changes between hydatidiform moles and villi
in whole genomes. Following NotI subtraction, MS-G-SH and
sequencing, three positive DNA clones were obtained. Moreover,
analysis of the CpG islands using searcher software indicated that
two positive CpG island segments, which had been modified by DNA
methylation, should be found by MS-G-SH. Following sequencing and
CpG island forecasting, the results of Blast tool analysis revealed
that these nucleic acid sequences had high similarities to the
secondary exon of the human IGF2 gene and the primary exon of human
TGF-β. According to these results, the TGF-β and IGF2 genes had
specific modifications of methylation that differed between
hydatidiform mole and villi. In addition, MS-PCR analysis indicated
that specific modification of DNA methylation existed in the TGF-β
and IGF2 exons of different samples. Furthermore, qRT-PCR showed
that when the DNA methylation of TGF-β and IGF2 differed, their
mRNA expression levels were affected. In conclusion, MS-G-SH is a
useful method for the genome-wide screening of deleted, amplified
and methylated NotI sites. This approach of screening and
analyzing DNA methylation patterns based on the altered composition
of PCR products with bisulfite treatment should facilitate future
studies of methylation.
Acknowledgements
This study was supported by a grant from the
Shanghai Municipal Health Bureau Fund for Young Scholars (no.
2008Y009) to Gang Zou, the PhD Programs Foundation of the Ministry
of Education of China (20110072110005) and the National Natural
Science Foundation of China (General Program; 30972823) to Tony
Duan.
References
|
1
|
Hales BF, Grenier L, Lalancette C and
Robaire B: Epigenetic programming: from gametes to blastocyst.
Birth Defects Res A Clin Mol Teratol. 91:652–665. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen ZX and Riggs AD: DNA methylation and
demethylation in mammals. J Biol Chem. 286:18347–18353. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar
|
|
4
|
Tomlinson IP, Webb E, Carvajal-Carmona L,
et al: A genome-wide association study identifies colorectal cancer
susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet.
40:623–630. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jaeger E, Webb E, Howarth K, et al: Common
genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3
influence colorectal cancer risk. Nat Genet. 40:26–28. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Houlston RS, Webb E, Broderick P, et al:
Meta-analysis of genome-wide association data identifies four new
susceptibility loci for colorectal cancer. Nat Genet. 40:1426–1435.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khandige S, Shanbhogue VV, Chakrabarty S
and Kapettu S: Methylation markers: a potential force driving
cancer diagnostics forward. Oncol Res. 19:105–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thapa K, Shrestha M, Sharma S and Pandey
S: Trend of complete hydatidiform mole. JNMA J Nepal Med Assoc.
49:10–13. 2010.PubMed/NCBI
|
|
9
|
Hayward BE, De Vos M, Talati N, et al:
Genetic and epigenetic analysis of recurrent hydatidiform mole. Hum
Mutat. 30:E629–E639. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
El-Maarri O, Seoud M, Coullin P, et al:
Maternal alleles acquiring paternal methylation patterns in
biparental complete hydatidiform moles. Hum Mol Genet.
12:1405–1413. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li AS, Siu MK, Zhang H, et al:
Hypermethylation of SOX2 gene in hydatidiform mole and
choriocarcinoma. Reprod Sci. 15:735–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Protopopov A, Wang F, et al: NotI
subtraction and NotI-specific microarrays to detect copy number and
methylation changes in whole genomes. Proc Natl Acad Sci USA.
99:10724–10729. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu T, Cheng W, Guo L, et al: Human
amniotic epithelial cell feeder layers maintain mouse embryonic
stem cell pluripotency via epigenetic regulation of the c-Myc
promoter. Acta Biochim Biophys Sin. 42:109–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paoloni-Giacobino A: Epigenetics in
reproductive medicine. Pediatr Res. 61:51R–57R. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu T, Zou G, Gao Y, et al: High
efficiency of reprogramming CD34+ cells derived from human amniotic
fluid into induced pluripotent stem cells with Oct4. Stem Cells
Dev. 21:2322–2332. 2012.
|
|
17
|
Bianco T, Hussey D and Dobrovic A:
Methylation-sensitive, single-strand conformation analysis
(MS-SSCA): a rapid method to screen for and analyze methylation.
Hum Mutat. 14:289–293. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bird A: Does DNA methylation control
transposition of selfish elements in the germline? Trends Genet.
13:469–472. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bird AP: Gene number, noise reduction and
biological complexity. Trends Genet. 11:94–100. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kashuba VI, Gizatullin RZ, Protopopov AI,
et al: Analysis of NotI linking clones isolated from human
chromosome 3 specific libraries. Gene. 239:259–271. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zabarovsky ER, Allikmets R, Kholodnyuk I,
et al: Construction of representative NotI linking libraries
specific for the total human genome and for human chromosome 3.
Genomics. 20:312–316. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zabarovsky ER, Gizatullin R, Podowski RM,
et al: NotI clones in the analysis of the human genome. Nucleic
Acids Res. 28:1635–1639. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zabarovsky ER, Kashuba VI, Kholodbnyuk ID,
Zabarovska VI, Stanbridge EJ and Klein G: Rapid mapping of NotI
linking clones with differential hybridization and Alu-PCR.
Genomics. 21:486–489. 1994. View Article : Google Scholar : PubMed/NCBI
|