Introduction
Dysregulated glucose metabolism underlies a variety
of pathologies leading to impairments in multiple types of cells
including neurons (1). Recently,
emerging evidence has accentuated the impact of high blood glucose
on neuronal function in vitro and in vivo suggesting
the correlation between glucose dysmetabolism and diverse types of
brain diseases including stroke and neurodegenerative diseases
(1,2). Mitochondria are the main
glucose-consuming sites in neurons and produce ATP by glycolysis
through oxidation phosphorylation. Previous studies have revealed
that high blood glucose substantially encumbers mitochondrial
function and results in mitochondria-dependent apoptosis suggesting
mitochondrial pathology stands in the nexus of glucose
dysmetabolism and cell injuries (3,4).
Among the many glucose dysmetabolism-induced mitochondrial
dysfunctions, increased mitochondria oxidative stress is a
predominant mitochondrial pathology and occurs early in response to
high-glucose insult (5).
Correspondingly, it has been widely reported that excessive
reactive oxygen species (ROS) plays a key role in the pathogenesis
of high glucose-associated pathological conditions such as
neurodegeneration and diabetes (5,6).
Oxidative stress is a strong inducer of the mitogen-activated
protein kinase P38 (P38 MAPK) activation (7) and the activation of P38 MAPK
signaling pathway is another prominent change in high
glucose-exposed cells. The excessive activation of P38 MAPK is a
critical mechanism underlying high glucose-induced cellular
injuries eventually leading to apoptosis (8,9).
Thus, elimination of high glucose-induced mitochondrial ROS may
have the potential to attenuate P38 MAPK signaling
disturbances.
The SS31 peptide (H-D-Arg-Dmt-Lys-Phe-NH2) is a
novel cell permeable antioxidant specifically targeted to
mitochondria and subsequently eliminates mitochondrial reactive
oxygen species (ROS) and preserves ATP production against oxidizing
insults (10,11). Yet, to the best of our knowledge,
the effect of the SS31 peptide on P38 MAPK signaling perturbation
in high glucose-insulted neurons has not yet been elucidated. In
the present study, using a neuron-like cell line (human
neuroblastoma SH-SY5Y) we demonstrated that the use of the SS31
peptide substantially suppressed high glucose-instigated
mitochondrial oxidative stress and P38 MAPK activation in
neuroblastoma cells which serves as evidence of the potential
application of mitochondria-targeted antioxidants in the treatment
of glucose dysmetabolism-induced neuronal stresses.
Materials and methods
Cell culture and treatment
Human neuroblastoma cells (SH-SY5Y) were obtained
from the American Type Culture Collection (ATCC). Cells were
cultured in DMEM (25 mM glucose, Gibco, USA) with 5% fetal bovine
serum (Gibco), 2 mM L-glutamine, 100 U/ml penicillin in a 95% air,
5% CO2 atmosphere. Cells were starved 12 h before the
experiments by replacing the medium with OptiMEM (Gibco) without
fetal bovine serum, followed by treatment with D-glucose (Sigma) as
vehicle (no additional glucose added) at indicated final
concentrations. The SS31 peptide (Stealth Peptide International,
China) at the indicated concentration was applied 30 min before the
addition of glucose. Antimycin A (Sigma) treatment was conducted at
50 μM on cells for 2 h. Carbonylcyanide
p-trifluoromethoxyphenylhydrazone (FCCP; Sigma) was used at 5 mM to
treat cells for 90 min.
Intracellular and intra-mitochondrial ROS
and mitochondrial membrane potential detection
Intracellular ROS was detected by using fluorescence
probe 5-(and 6-) chloromethyl-20, 70-dichlorodihydrofluorescein
diacetate (CM-DCF-DA; Invitrogen, USA) at 10 μM for 30 min, and
intra-mitochondrial ROS was visualized by coincubation of MitoSox
Red (Invitrogen) at 1 μM. To detect mitochondrial membrane
potential, tetramethylrhodamine methyl ester (TMRM; Invitrogen) was
used at 200 nM for 20 min followed by washing using culture medium.
Cells, after appropriate staining were observed under a Zeiss
Axiovert 200M inverted fluores,cence microscope with heating
control and CO2 chamber. The intensity of fluorescent
staining was calculated as (F1-F0). F1 is the intensity of the
staining and F0 is the intensity of the background.
Western blotting
The proper amount of cell extracts was loaded and
separated by SDS-PAGE and transferred to nitrocellulose membranes
(Bio-Rad, USA). Immunoblotting was performed using antibodies
against phospho P38 (pT180/pY182, BD; 1:1,000), P38 (Cell
Signaling, USA; 1:3,000) or β-actin (Sigma; 1:5,000) overnight at
4˚C. Immunoreactive bands were visualized using the ECL system
(Fisher Scientific, USA).
TUNEL assay
Cell apoptosis was measured by terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method
using the TUNEL Assay kit (Roche, Germany) following the
manufacturer's instructions.
Statistics and data analysis
Statistical analysis was performed by one-way ANOVA
and post-hoc ANOVA analysis when appropriate using SPSS software.
The values are shown as mean ± SEM. Significance was defined as
P<0.05. Analysis of immunoblotting and cell staining was
conducted using NIH ImageJ software (1.62).
Results
High glucose induces P38 MAPK activation
in SH-SY5Y cells in a dose-dependent manner
Cells were exposed to vehicle, 60, 90, 120 and 150
mM glucose for 90 min after starvation and then subjected to
immunoblotting to detect the levels of phospho P38 and total P38
MAPK. β-actin was adopted as the loading control to verify that the
same amount of proteins was loaded in each well. Our data showed
that the P38 phosphorylation level was increased in a
dose-dependent manner with increased glucose concentration. Glucose
treatment (60 mM) induced a mild increase in P38 phosphorylation as
compared with the vehicle group (Fig.
1A, P>0.05); whereas glucose at concentrations of 90 mM or
higher significantly instigated P38 phosphorylation in the treated
cells (Fig. 1A, P<0.05 vs.
control group). In contrast, the level of total P38 was not changed
in any group by comparison with β-actin suggesting that the
increased P38 phosphorylation in the glucose treated-groups was not
due to an increased level of total P38.
SS31 peptide protects mitochondrial
membrane potential against high-glucose insult
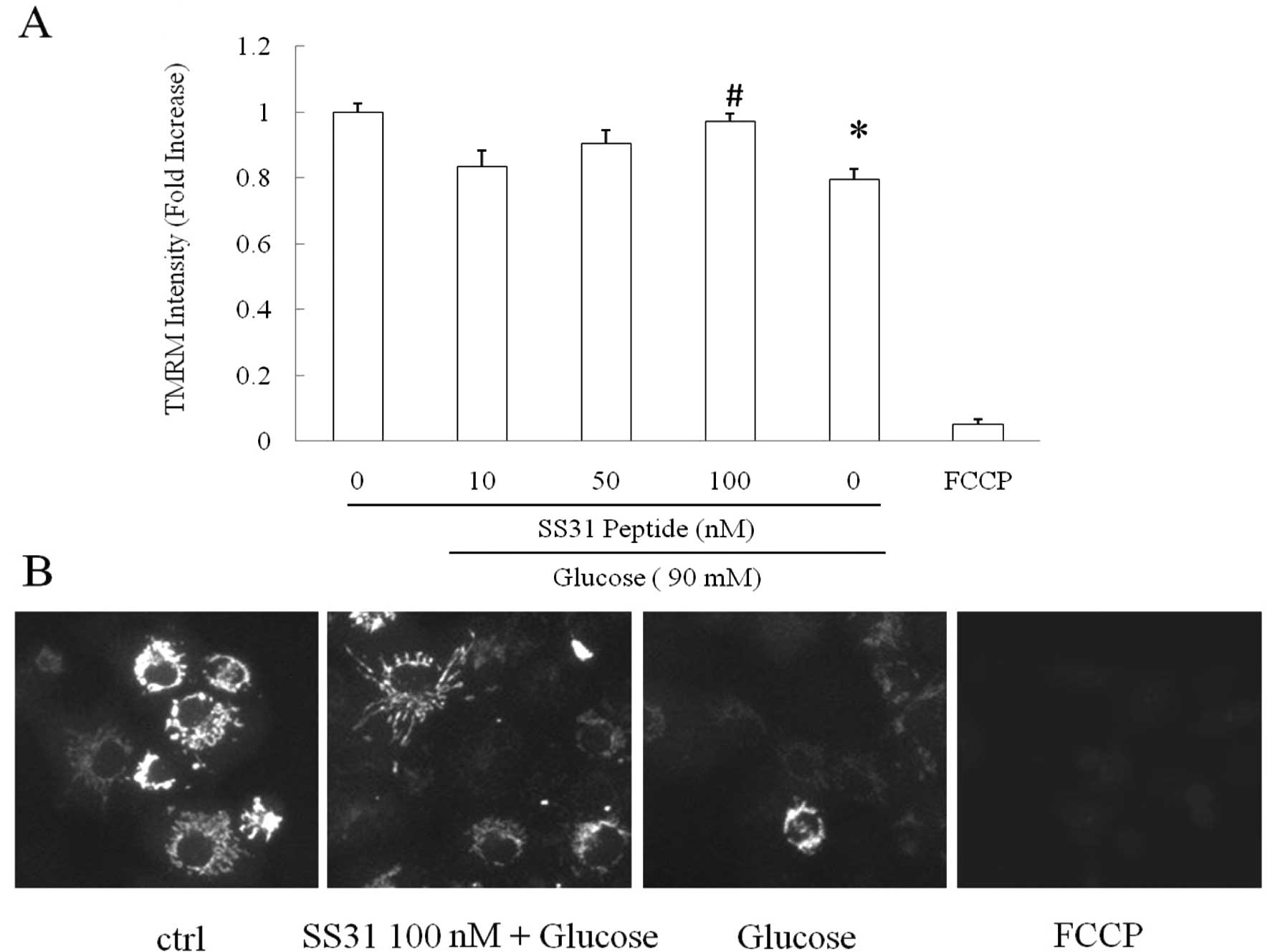
Mitochondrial membrane potential is a critical
indicator of mitochondrial function, and decreased mitochondrial
membrane potential is closely associated with elevated
mitochondrial ROS production. Previous studies have shown that the
SS31 peptide confers substantial protection on mitochondrial
membrane potential against multiple insults (3,12,13).
We next studied whether the SS31 peptide preserves mitochondrial
membrane potential against high glucose in SH-SY5Y cells. FCCP, a
strong mitochondrial uncoupler, was employed to treat cells at 5 mM
as a positive control. High glucose substantially decreased
mitochondrial membrane potential to 79.5% in comparison to the
vehicle group (Fig. 2,
P<0.0001); whereas the adoption of the SS31 peptide demonstrated
a dose-dependent rescue of the high glucose-induced mitochondrial
membrane potential collapse. Of note, the SS31 peptide at the
concentration of 100 nM exhibited significant restoration of high
glucose-insulted mitochondrial membrane potential decrease almost
to the same level as that in the vehicle group (Fig. 2, vs. high glucose-treated group;
P<0.0001); while the SS31 peptide at 50 nM also showed partial
recovery as compare to the high glucose-treated group (Fig. 2A, P<0.05). FCCP-exposed cells
demonstrated significantly lower mitochondrial membrane potential
indicating the specificity of TMRM as a mitochondrial membrane
potential indicator.
SS31 peptide suppresses high
glucose-induced intracellular and intra-mitochondrial ROS
production
Mitochondrial membrane potential collapse is closely
associated with mitochondrial ROS production. As the SS31 peptide
at 100 nM significantly restored mitochondrial membrane potential
in SH-SY5Y cells against high-glucose insult, we next observed the
impact of the SS31 peptide at 100 nM on high glucose-induced
cellular and mitochondrial ROS production. We compared the
intracellular and intra-mitochondria ROS levels between the
vehicle- and glucose-treated groups. Intracellular ROS was detected
using DCF-DA and intra-mitochondrial ROS was shown as the increased
intensity of MitoSox Red, a specific mitochondrial ROS indicator.
The data showed that cells exposed to 90 mM glucose for 90 min
underwent significantly increased intra-mitochondrial and
intracellular ROS levels by 40.5% (Fig. 3A and C, P<0.0001) and 45.5%
(Fig. 3B and C, P=0.0047),
respectively, as compared with the vehicle group. In sharp
contrast, the addition of the SS31 peptide significantly suppressed
the high glucose-induced increase in intracellular and
intra-mitochondrial ROS levels. In the SS31 peptide-treated group,
the intra-mitochondrial ROS level was reduced by 28.0% (Fig. 3A and C, P=0.0008) while
intracellular ROS level was decreased by 26.6% (Fig. 3B and C, P=0.0359) in comparison
with the high glucose-treated group implicating the effect of SS31
peptide application on high glucose-instigated ROS generation.
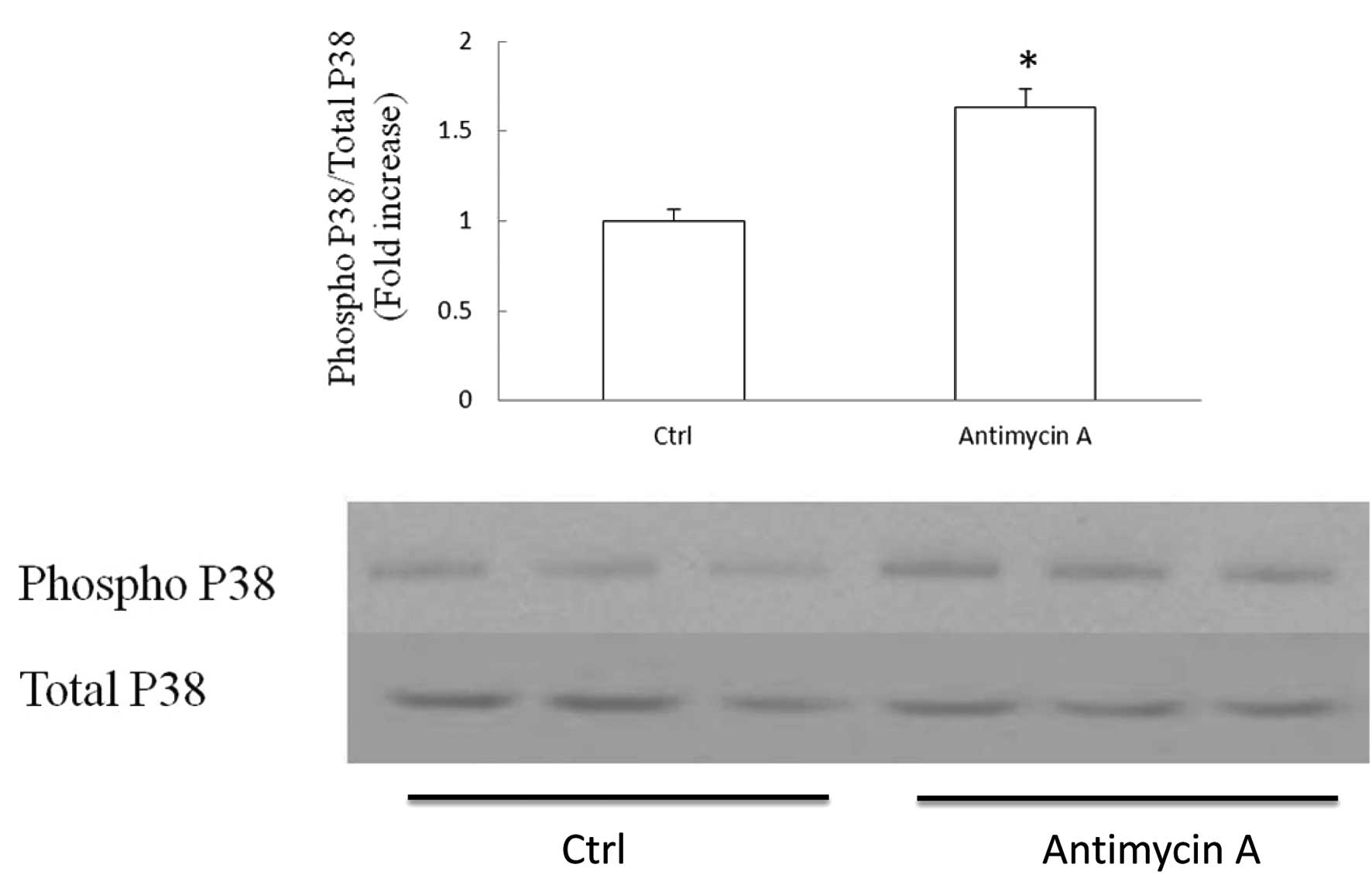
Elevated mitochondrial ROS production
instigates P38 MAPK activation in SH-SY5Y cells
Oxidative stress is a known causative factor of P38
activation. Mitochondria are the major sites of ROS production. In
order to examine whether P38 activation is related to mitochondrial
ROS production in SH-SY5Y cells, we applied antimycin A to cultured
cells. Antimycin A is the inhibitor of mitochondrial complex III
and is a strong inducer of mitochondrial ROS production. Cells were
exposed to antimycin A at 50 μM for 2 h and subjected to
immunoblotting to detect the P38 phosphorylation level. The ratio
of phospho P38 to total P38 was employed as the manifestation of
P38 activation. Our data showed that antimycin A induced a 63%
increase in P38 activation level in comparison to the control
(Fig. 4, P<0.05). The result
indicates that mitochondrial ROS production/release is associated
with P38 activation in SH-SY5Y cells.
SS31 peptide inhibits high
glucose-induced P38 phosphorylation in SH-SY5Y cells in a
dose-dependent manner
Finally, we determined whether suppressed
mitochondrial ROS subsequently inhibits high glucose-instigated P38
activation. We employed the SS31 peptide at 0, 10, 50 100 and 200
nM, respectively. Cells were exposed to the SS31 peptide at
indicated concentrations for 30 min followed by incubation with
glucose at 90 mM for 90 min. The coincubation of the SS31 peptide
with high glucose markedly attenuated high glucose-induced P38
activation in a dose-dependent manner. As shown in Fig. 5, high-glucose treatment increased
the P38 phosphorylation level to 2.33±0.14-fold as compared with
the vehicle group (Fig. 5A,
P<0.05). In sharp contrast, the application of the SS31 peptide
at 100 and 200 nM substantially decreased P38 phosphorylation to a
similar level in comparison with that in the vehicle control
(P>0.05 vs. vehicle group), which was significantly lower than
that in the high glucose-treated group (Fig. 5A, P<0.05). Although the SS31
peptide at 10 nM exhibited limited inhibition on high
glucose-induced P38 activation, the SS31 peptide at 50 nM
demonstrated partial protection on high glucose-induced P38
activation (Fig. 5A, P<0.05).
The dose-dependent inhibition of the SS31 peptide on
glucose-induced P38 activation was in correlation with the impact
of the SS31 peptide on high glucose-instigated mitochondrial
membrane potential and ROS production perturbations. The addition
of P38 inhibitor SB203580 (200 nM) significantly suppressed high
glucose-induced P38 activation (Fig.
5A, P<0.05 vs. high glucose-treated group). Thus, our data
indicate a protective effect of the SS31 peptide on P38 activation
in SH-SY5Y cells against high-glucose exposure.
SS31 peptide attenuates high
glucose-induced SH-SY5Y cell apoptosis
P38 activation has been implicated to be involved in
apoptosis in response to cell injuries. We then assessed the
protection of the SS31 peptide on apoptosis in the face of
high-glucose insult. SH-SY5Y cells were exposed to 90 mM glucose in
the presence or absence of 100 nM SS31 peptide for 24 h and then
subjected to TUNEL assay. Our data showed that high glucose-induced
a 2.6-fold increase in the percentage of apoptotic cells while the
application of the SS31 peptide significantly ameliorated the high
glucose-induced apoptosis (Fig. 6,
P<0.05 vs. other groups). The SS31 peptide alone did not have a
significant effect on cell apoptosis.
Discussion
Hyperglycemia is a potential culprit underlying
peripheral neuron changes in diabetes and neurodegeneration in the
central nervous system (CNS) (6,14).
The P38 MAPK signaling pathway plays a key role in controlling cell
proliferation while excessive P38 activation leads to
mitochondria-dependent apoptosis. It should be noted that P38 MAPK
signaling activation is a predominant cell change in response to
high-glucose insult and is significantly involved in high
glucose-related cell injuries including decreased mitochondrial
function and mitochondria-dependent apoptosis in the pathogenesis
of hyperglycemia-associated diseases (9,15).
Previous studies have shown that mitochondrial ROS is an initiator
of P38 activation in cardiomyocytes and neurons under hypoxia and
hyperglycemic conditions (16,17).
The elimination of excessive cellular ROS has the potential to
protect against high glucose-induced P38 activation. Indeed,
various ROS scavengers such as VitE, SOD and lipid acid have been
applied to treat hyperglycemia-associated cell stresses. However,
the applications of these antioxidants are limited by multiple
drawbacks such as high drug dose and various side effects (18–20).
The SS31 peptide is a novel antioxidant specifically targeted to
mitochondria and efficiently quenches mitochondrial ROS in many
pathological conditions. Importantly, its relatively
small-molecular weight and lipid solubility enable the peptide to
rapidly penetrate cells (21). Our
result showed that the addition of the SS31 peptide significantly
reversed the high glucose-induced mitochondrial ROS elevation and
mitochondrial membrane potential collapse. These findings are in
agreement with many previous studies on the pharmaceutical effects
of the SS31 peptide (10,13). Furthermore, by eliminating
mitochondrial ROS and protecting mitochondrial membrane potential,
P38 activation as well as apoptosis in high glucose-treated SH-SY5Y
cells was substantially suppressed by the application of the SS31
peptide. The SS31 peptide protects cells from high-glucose insult.
Notably, we adopted a neuron-like cell line, SH-SY5Y which
demonstrates neuron biological features. To the best of our
knowledge, to date, reports are limited on the effect of SS31
peptide against high glucose-induced mitochondrial ROS as well as
neuronal dysfunctions. These results serve as important evidence
for the application of this peptide in preventing neuronal stresses
and injury related to hyperglycemia.
Collectively, we demonstrated that the nascent
mitochondria-targeted antioxidant, SS31 peptide, substantially
rescues high glucose-associated mitochondrial dysfunction and P38
activation in SH-SY5Y cells. This provides evidence that the SS31
peptide is a promising treatment strategy for rescuing neuronal
function from hyperglycemia. Moreover, the small size of the SS31
peptide and its lipid solubility substantially enable the use of
the SS31 peptide in treating hyperglycemia-instigated neuronal
stresses in the CNS.
Acknowledgements
This study was supported by a grant from the Science
Foundation of Shandong Province (Grant No. Q2006C15), China.
References
|
1
|
F DasN DeyB VenkatesanBS KasinathN
Ghosh-ChoudhuryGG ChoudhuryHigh-glucose upregulation of early-onset
Parkinson's disease protein DJ-1 integrates the PRAS40/TORC1 axis
to mesangial cell hypertrophyCell Signal2313111319201121426932
|
|
2
|
T MatsuzakiK SasakiY TanizakiInsulin
resistance is associated with the pathology of Alzheimer disease:
the Hisayama
studyNeurology75764770201010.1212/WNL.0b013e3181eee25f20739649
|
|
3
|
J LiX ChenW XiaoMitochondria-targeted
antioxidant peptide SS31 attenuates high-glucose-induced injury on
human retinal endothelial cellsBiochem Biophys Res
Commun404349356201110.1016/j.bbrc.2010.11.12221134355
|
|
4
|
M SchiffS LoublierA CoulibalyP BenitHO de
BaulnyP RustinMitochondria and diabetes mellitus: untangling a
conflictive relationship?J Inher Metab
Dis32684698200910.1007/s10545-009-1263-019821144
|
|
5
|
JA HerleinBD FinkWI SivitzSuperoxide
production by mitochondria of insulin-sensitive tissues:
mechanistic differences and effect of early
diabetesMetabolism59247257201010.1016/j.metabol.2009.07.02119765776
|
|
6
|
HL WangAH ChouAS WuPARK6 PINK1 mutants are
defective in maintaining mitochondrial membrane potential and
inhibiting ROS formation of substantia nigra dopaminergic
neuronsBiochimic Biophys
Acta1812674684201110.1016/j.bbadis.2011.03.00721421046
|
|
7
|
J RavindranN GuptaM AgrawalAS Bala
BhaskarPV Lakshmana RaoModulation of ROS/MAPK signaling pathways by
okadaic acid leads to cell death via mitochondrial-mediated
caspase-dependent
mechanismApoptosis16145161201110.1007/s10495-010-0554-021082355
|
|
8
|
N BegumL RagoliaHigh-glucose and insulin
inhibit VSMC MKP-1 expression by blocking iNOS via P38 MAPK
activationAm J Physiol Cell Physiol278C81C91200010644515
|
|
9
|
Y RenY ShiY WangP38 MAPK pathway is
involved in high-glucose-induced thioredoxin interacting protein
induction in mouse mesangial cellsFEBS
Lett58434803485201010.1016/j.febslet.2010.07.01020624390
|
|
10
|
EA CarterAA BonabJ GovermanEvaluation of
the antioxidant peptide SS31 for treatment of burn-induced insulin
resistanceInt J Mol Med28589594201121805045
|
|
11
|
TP ReddyM ManczakMJ CalkinsToxicity of
neurons treated with herbicides and neuroprotection by
mitochondria-targeted antioxidant SS31Inter J Environ Res Public
Health8203221201110.3390/ijerph801020321318024
|
|
12
|
DC AnderssonJ FauconnierT
YamadaMitochondrial production of reactive oxygen species
contributes to the beta-adrenergic stimulation of mouse
cardiomycytesJ
Physiol58917911801201110.1113/jphysiol.2010.20283821486840
|
|
13
|
M ManczakP MaoMJ
CalkinsMitochondria-targeted antioxidants protect against
amyloid-beta toxicity in Alzheimer's disease neuronsJ Alzheimers
Dis20Suppl 2S609S6312010
|
|
14
|
C TothV BrusseeC ChengDW ZochodneDiabetes
mellitus and the sensory neuronJ Neuropathol Exp
Neurol63561573200415217085
|
|
15
|
WA WilmerCL DixonC HebertChronic exposure
of human mesangial cells to high-glucose environments activates the
P38 MAPK pathwayKidney
Int60858871200110.1046/j.1523-1755.2001.060003858.x11532081
|
|
16
|
A KuliszN ChenNS ChandelZ ShaoPT
SchumackerMitochondrial ROS initiate phosphorylation of p38 MAP
kinase during hypoxia in cardiomyocytesAm J Physiol Lung Cell Mol
Physiol282L1324L1329200210.1152/ajplung.00326.200112003789
|
|
17
|
R SharmaE BurasT TerashimaHyperglycemia
induces oxidative stress and impairs axonal transport rates in
micePloS One5e13463201010.1371/journal.pone.001346320976160
|
|
18
|
M BrownleeThe pathobiology of diabetic
complications: a unifying
mechanismDiabetes5416151625200510.2337/diabetes.54.6.161515919781
|
|
19
|
F Guerrero-RomeroM
Rodriguez-MoranComplementary therapies for diabetes: the case for
chromium, magnesium, and antioxidantsArch Med
Res36250257200510.1016/j.arcmed.2005.01.00415925015
|
|
20
|
AM VincentJW RussellP LowEL
FeldmanOxidative stress in the pathogenesis of diabetic
neuropathyEndocr Rev25612628200410.1210/er.2003-001915294884
|
|
21
|
S ChoHH SzetoE KimH KimAT TolhurstJT
PintoA novel cell-permeable antioxidant peptide, SS31, attenuates
ischemic brain injury by down-regulating CD36J Biol
Chem28246344642200710.1074/jbc.M60938820017178711
|