Introduction
Nicotine, a major component of cigarette smoke,
unequivocally, has positive effects on illnesses as diverse as
neurodegenerative diseases, ulcerative colitis and Tourette
syndrome (1–3). Although the expression of nicotinic
acetylcholine receptor (nACh) has been demonstrated in many types
of non-neuronal cells such as dendritic cells (DCs), epithelial and
endothelial cells (4), the effect
of nicotine on immune cells is incompletely characterized. Aicher
et al found that nicotine activates DCs and augments their
capacity to stimulate T cell proliferation and cytokine secretion,
which may contribute to the progression of atherosclerotic lesions
(5). Our previous studies further
demonstrated that nicotine has stimulatory effects on immature
dendritic cells (imDCs), which reveal anti-tumor effects on
lymphoma development (6), lung and
liver cancer (7). Nouri-Shirazi
et al reported that nicotine exerts immunosuppressive
effects on immune surveillance through functional impairment of the
DC system (8). In parallel with
differential expression of costimulatory molecules CD80 and CD86
and lack of IL-12, nicotine-stimulated DCs displayed profoundly
reduced Th1-promoting capacity (4), which recently confirmed that the
presence of nicotine in the microenvironment promoted the
development of mouse bone marrow-derived DC precursors into DCs
with a semi-mature phenotype revealed by higher expression of
costimulatory molecules CD80, CD86, CD40 and MHC II (9). Investigators have shown that nicotine
promotes immune cell activation (5–7),
whereas others have suggested that nicotine may have
immunosuppressive effects on DCs (4,8,9).
Since the biological effect of nicotine on lymphocytes is dependent
on dose and duration of exposure (10), the controversial effects of
nicotine on DCs may be attributed to differences in experimental
design, species, duration of exposure, particularly the nicotine
concentration used in these experiments. Hence, further studies are
needed to explore the factors which dictate the effects of nicotine
on DCs.
In the present study, we first found that nicotine
treatment up-regulated CD11c expression on imDCs in the absence of
LPS, and secondly that lower and higher doses of nicotine used in
previous reports up- or down-regulated the expression of
co-stimulatory molecules on imDCs. Co-administration of LPS and
nicotine revealed differential effects of expression of the
co-stimulatory molecules on imDCs. Thirdly and importantly, lower
doses of nicotine treatment did not augment expression of CD80,
CD86, CD40 and CD54 molecules on mature DCs. Fourthly and
interestingly, high doses of nicotine (more than 165 μg/ml)
revealed pro-apoptotic activity and lower doses of nicotine
(16.5–0.165 ng/ml) achieved an anti-apoptotic effect on imDCs.
These data presented here indicate that the controversial effects
of nicotine on DCs may be due to the nicotinic environment and the
dose of nicotine used.
Materials and methods
Reagents
Nicotine and lipopolysaccharides (LPS) were obtained
from Sigma-Aldrich (St. Louis, MI, USA). Mouse GM-CSF and IL-4 were
obtained from R&D (Minneapolis, MN, USA).
Fluorescent-conjugated antibodies were from eBioscience (San Diego,
CA, USA). Annexin-V apoptosis detection kit was obtained from
Promega (Madison, WI, USA). RPMI-1640 medium, Dulbecco's modified
Eagle's medium (DMEM) and fetal bovine serum were purchased from
Hyclone (Logan, UT, USA).
Animals
Pathogen-free C57BL/6 mice (female, 6–8 weeks old)
were purchased from Shanghai Laboratory Animal Center of the
Chinese Academy of Sciences (China) and kept at the Animal Center
of Xiamen University. All animal studies were approved by the
Review Board of the Medical College of Xiamen University.
Bone marrow-derived murine DCs
Bone marrow-derived DCs were prepared as previously
described (11). Briefly, bone
marrow mononuclear cells were prepared from bone marrow suspensions
by depletion of red cells, and were then cultured at a density of
1×106 cells/ml in RPMI-1640 medium with 10 ng/ml of
GM-CSF and 1 ng/ml of IL-4. Non-adherent cells were gently washed
out on day 4 of culture; the remaining loosely adherent clusters
were used as imDCs. Both imDCs and mature (ma)DCs (1×106
cells) were firstly starved in RPMI-1640 medium + 0.5% FCS for 6 h
and exposed to nicotine (16.5 ng/ml) for 12 h. After washings, the
cells were used as nicotine-treated DCs. imDCs were cultured for a
further 4 days in the presence of 10 ng/ml LPS and used as
maDCs.
Flow cytometric measurement
Expression of cell surface molecules was determine
by flow cytometry according to the methods described previously
(11). Before staining with
relevant Abs, imDCs were incubated for 15 min at 4˚C with an
antibody to CD16/CD32 at a concentration of 1 μg per
1×106 cells for blockade of Fc receptors. Staining was
performed on ice for 30 min and then cells were washed with
ice-cold PBS, containing 0.1% NaN3 and 0.5% BSA. Flow
cytometry was carried out with FACSCalibur, and data were analyzed
with CellQuest software.
Cell apoptosis assay
Cell apoptosis assay was determined by flow
cytometry according to the method described previously (5). For detection of cell apoptosis, DCs
were collected from PBS or nicotine-treated imDCs. Cell suspension
was washed in PBS and resuspended in binding buffer containing
Annexin V-FITC and propidium iodide (PI) for 20 min at room
temperature. The samples were analyzed on FACSCalibur and data were
analyzed with CellQuest software.
Statistical analysis
All data were expressed as the average of
experimental data points, and standard error means were determined
using the calculated standard deviation of a data set divided by
the number of data points within the data set. Statistical
significance was tested using the Student's t-test and one-way
ANOVA test by Prism software. Differences were considered to be
statistically significant at p<0.05.
Results
Nicotine treatment promotes
differentiation of DC precursors into DCs
When cultured in the presence of GM-CSF with IL-4,
DC precursors in the bone marrow differentiated into imDCs,
expressing CD11c (12). In order
to explore the role of nicotine on DC differentiation, imDCs
derived from murine bone marrow were stimulated with nicotine, and
the morphology and expression of CD11c were observed by inverted
microscopy and flow cytometry, respectively. The results showed
that imDCs induced on day 6 grew more branched projections compared
to those on day 4, and the expression of CD11c was also increased
from 11.19 to 25.00% (Fig. 1A).
When imDCs of day 4 were stimulated by nicotine (16.5 ng/ml), more
branched projections on imDCs were observed, and the expression of
CD11c was up-regulated from 11.19 to 25.68% (Fig. 1A). Compared to imDCs on day 6,
imDCs on day 4 stimulated with nicotine had more CD11c molecular
expression (Fig. 1B, p=0.0002,
imDC day 4 vs. imDC day 4 + Ni; p=0.0005, imDC day 4 vs. imDC day
6; p=0.0013, imDC day 4 + Ni vs. imDC day 4; Fig. 1C, p=0.0009, imDC day 4 vs. imDC day
4 + Ni; p<0.0001, imDC day 4 vs. imDC day 6; p=0.0154, imDC day
4 + Ni vs. imDC day 6). Since CD11c is a marker of DCs, the
up-regulation of CD11c by nicotine indicated that nicotine enhanced
DC differentiation from DC precursors.
Lower doses of nicotine up-regulate the
expression of co-stimulatory molecules on imDCs
Several reports have described the controversial
effects of nicotine on the expression of DC co-stimulatory
molecules (4–9,13).
To investigate the effects of nicotine on the expression of
co-stimulatory molecules in DCs, imDCs on day 4 were treated with
different doses of nicotine (16.5 ng/ml, 25 and 200 μg/ml), and the
expression levels of CD80, CD86, CD40 and CD54 were determined by
flow cytometry. The results showed that 16.5 ng/ml of nicotine
stimulation obviously increased CD80, CD40 and CD54 molecular
expression but decreased CD86 expression on imDCs (Fig. 2B, p=0.0256, imDC control vs. Ni
16.5 ng/ml; Fig. 2D, p=0.0098,
imDC control vs. Ni 16.5 ng/ml; Fig.
2F, p=0.0240, imDC control vs. Ni 16.5 ng/ml; Fig. 2H, p=0.0013, imDC control vs. Ni
16.5 ng/ml); 25 μg/ml of nicotine treatment also up-regulated CD86
and CD40 molecular expression (Fig.
2D, p=0.0011, imDC control vs. Ni 25 μg/ml; Fig. 2F, p=0.0459, imDC control vs. Ni 25
μg/ml). When 200 μg/ml of nicotine was used to stimulate imDCs,
down-regulation of both CD80 and CD54, as well as up-regulation of
CD40 were observed (Fig. 2B,
p=0.0004, Ni 16.5 ng/ml vs. Ni 200 μg/ml; Fig. 2D, p=0.0010, Ni 25 μg/ml vs. Ni 200
μg/ml; Fig. 2F, p<0.0001, imDC
control vs. Ni 200 μg/ml; p<0.0010, Ni 25 μg/ml vs. Ni 200
μg/ml; Fig. 2H, p<0.0001, imDC
control vs. Ni 200 μg/ml).
 | Figure 2Lower doses of nicotine up-regulate
the expression of co-stimulatory molecules on imDCs. imDCs on day 4
induced from murine bone marrow were treated with 16.5 ng/ml, 25
and 200 μg/ml of nicotine for 12 h, and the expression of CD80,
CD86, CD40 and CD54 on DCs was determined by flow cytometry. (A)
Histographic presentation of CD80 expression on imDCs. (B) CD80
expression on imDCs (p=0.0256, imDC control vs. Ni 16.5 ng/ml;
p=0.0004, Ni 16.5 ng/ml vs. Ni 200 μg/ml). (C) Histographic
presentation of CD86 expression on imDCs. (D) CD86 expression on
imDCs (p=0.0098, imDC control vs. Ni 16.5 ng/ml; p=0.0011, imDC
control vs. Ni 25 μg/ml; p=0.0437, imDC control vs. Ni 200 μg/ml;
p=0.0010, Ni 25 μg/ml vs. 200 μg/ml). (E) Histographic presentation
of CD40 expression on imDCs. (F) CD40 expression on imDCs
(p=0.0240, imDC control vs. Ni 16.5 ng/ml; p=0.0459, imDC control
vs. Ni 25 μg/ml; p<0.0001, imDC control vs. Ni 200 μg/ml;
p<0.0010, Ni 25 μg/ml vs. Ni 200 μg/ml). (G) Histographic
presentation of CD54 expression on imDCs. (H) CD54 expression on
imDCs (p=0.0013, imDC control vs. Ni 16.5 ng/ml; p<0.0001, Ni
16.5 ng/ml vs. Ni 25 μg/ml; p<0.0001, imDC control vs. Ni 200
μg/ml). A representative flow cytometry analysis out of 3 was
shown; Student's t test. Ni, nicotine. |
Nicotine (16.5 ng/ml) and LPS
co-administration obviously up-regulate co-stimulatory molecules on
imDCs
LPS was found to promote DC maturation and to
up-regulate co-stimulatory molecules (9). Burgdorf et al reported that
LPS activates the TLR4 pathway and increase DC cross-presentation
(14). In order to explore the
effects of nicotine and LPS on co-stimulatory molecule expression,
imDCs on day 4 were stimulated with LPS and nicotine, and the
expression of CD80, CD86, CD40 and CD54 was determined by flow
cytometry. The results showed that, in the presence of LPS, 16.5
ng/ml nicotine obviously increased the expression of the
co-stimulatory molecules CD80, CD86, CD40 and CD54 on imDCs
(Fig. 3A, p<0.0001, imDC
control vs. Ni 16.5 ng/ml; p<0.0001, Ni 16.5 ng/ml vs. Ni 25
μg/ml; Fig. 3B, p<0.0001, imDC
control vs. Ni 16.5 ng/ml; p<0.0001, Ni 16.5 ng/ml vs. Ni 25
μg/ml; Fig. 3C, p<0.0001, imDC
control vs. Ni 16.5 ng/ml; p<0.0001, imDC control vs. Ni 25
μg/ml; Fig. 3D, p<0.0001, imDC
control vs. Ni 16.5 ng/ml; p<0.0001, imDC control vs. Ni 25
μg/ml). In contrast to 16.5 ng/ml of nicotine stimulation, 200
μg/ml of nicotine obviously decreased CD80, CD86 and CD54
expression; however, increased CD40 expression on imDCs in the
presence of LPS was noted (Fig.
3A, p<0.0001, imDC control vs. Ni 200 μg/ml; p<0.0001, Ni
25 μg/ml vs. Ni 200 μg/ml; Fig.
3B, p=0.0032, Ni 25 μg/ml vs. Ni 200 μg/ml; Fig. 3C, p=0.0004, imDC control vs. Ni 200
μg/ml; p=0.0440, Ni 16.5 ng/ml vs. Ni 200 μg/ml; p=0.0252, Ni 25
μg/ml vs. Ni 200 μg/ml. Fig. 3D,
p<0.0001, imDC control vs. Ni 200 μg/ml).
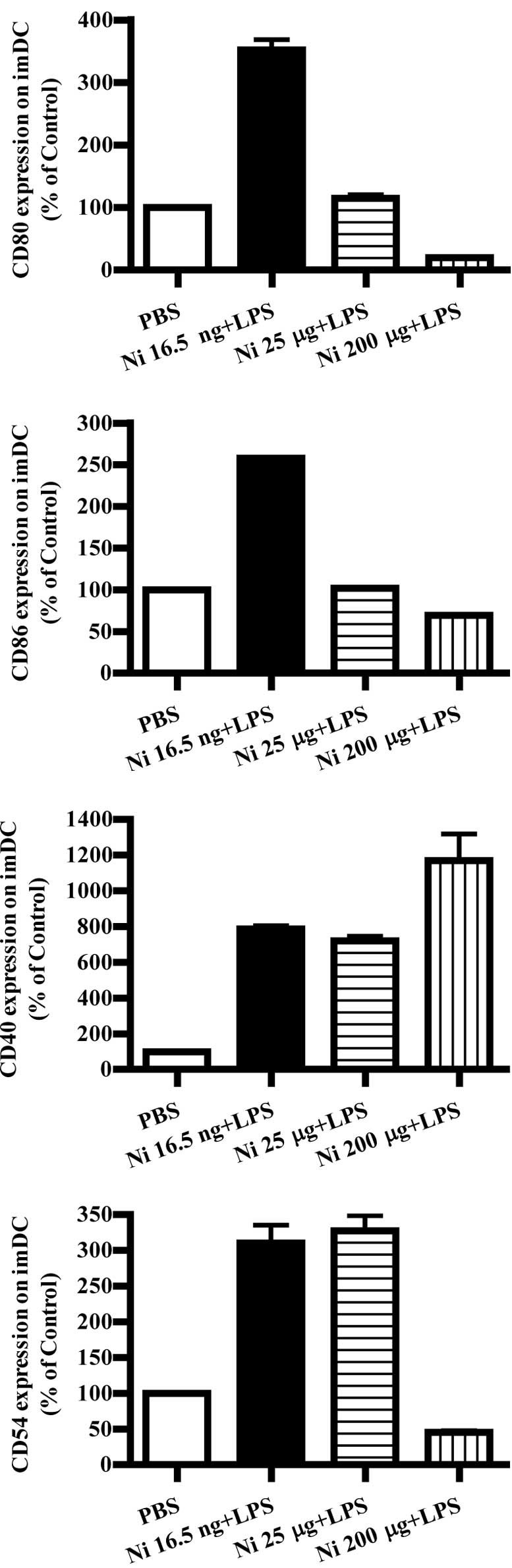 | Figure 3Lower doses of nicotine up-regulate
the expression of surface molecules on imDCs in the presence of
LPS. imDCs were treated with 16.5 ng/ml, 25 and 200 μg/ml of
nicotine in the presence of 100 ng/ml LPS for 12 h on day 4, and
then the expression levels of CD80, CD86, CD40 and CD54 on DCs were
determined by flow cytometry. (A) CD80 expression on imDCs with
different doses of nicotine and LPS stimulation (p<0.0001, imDC
control vs. Ni 16.5 ng/ml; p<0.0001, imDC control vs. Ni 200
μg/ml; p<0.0001, Ni 16.5 ng/ml vs. Ni 25 μg/ml; p<0.0001, Ni
25 μg/ml vs. Ni 200 μg/ml). (B) CD86 expression on imDCs with
different doses of nicotine and LPS stimulation (p<0.0001, imDC
control vs. Ni 16.5 ng/ml; p<0.0001, Ni 16.5 ng/ml vs. Ni 25
μg/ml; p=0.0032 and Ni 25 μg/ml vs. Ni 200 μg/ml). (C) CD40
expression on imDCs with different doses of nicotine and LPS
stimulation (p<0.0001, imDC control vs. Ni 16.5 ng/ml;
p<0.0001, imDC control vs. Ni 25 μg/ml; p=0.0004, imDC control
vs. Ni 200 μg/ml; p=0.0440, Ni 16.5 ng/ml vs. Ni 200 μg/ml;
p=0.0252, Ni 25 μg/ml vs. Ni 200 μg/ml). (D) CD54 expression on
imDCs with different doses of nicotine and LPS stimulation
(P<0.0001, imDC control vs. Ni 16.5 ng/ml; p<0.0001, imDC
control vs. Ni 25 μg/ml; p<0.0001, imDC control vs. Ni 200
μg/ml). A representative flow cytometric analysis out of 3 is
shown; Student's t test. Ni, nicotine. |
Nicotine has little effect on
co-stimulatory molecule expression in mature DCs
With DC maturation, the co-stimulatory molecules
were up-regulated accordingly. Mature immunogenic DCs were found to
induce Th1 and Th2 cell differentiation, and/or CTL priming,
depending on the nature of the maturation signal they received, as
well as the constraints imposed by ontogeny and/or environment
modifiers (10). Although nicotine
increased co-stimulatory molecule expression on imDCs, its effects
on co-stimulatory molecule expression of mature DCs is little
known. To investigate the effects of nicotine on mature DC
co-stimulatory molecule expression, DCs induced from murine bone
marrow were matured by LPS first and further stimulated with
nicotine. The results showed that, 16.5 ng/ml of nicotine
stimulation decreased the expression of CD80, CD86 and CD40 but
increased CD54 expression on mature DCs (Fig. 4B, p=0.0256, imDC control vs. Ni
16.5 ng/ml; Fig. 4D, p=0.0098,
imDC control vs. Ni 16.5 ng/ml; Fig.
4F, p=0.0240, imDC control vs. Ni 16.5 ng/ml; Fig. 4H, p=0.0013, imDC control vs. Ni
16.5 ng/ml). In contrast to 16.5 ng/ml of nicotine stimulation, 25
μg/ml nicotine stimulation obviously augmented the expression of
CD80 and CD86 on mature DCs (Fig.
4B, p=0.0004, Ni 16.5 ng/ml vs. Ni 200 μg/ml; Fig. 4D, p=0.0011, imDC control vs. Ni 25
μg/ml).
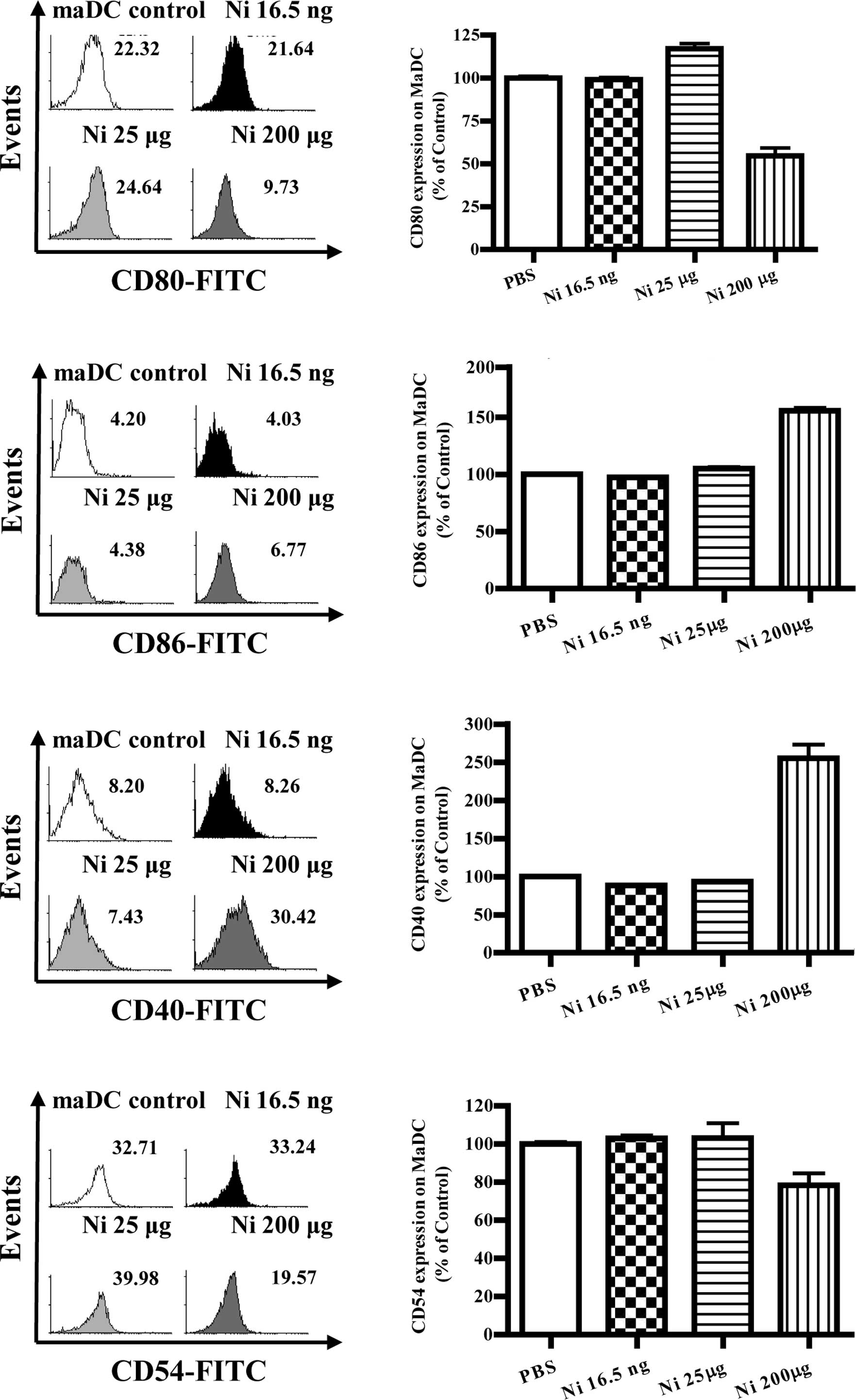 | Figure 4The doses of nicotine determine the
expression of surface molecules on maDCs. At day 4, imDCs induced
from murine bone marrow were treated with 10 ng/ml LPS for a
further 4 days and were considered as mature DCs (maDCs). maDCs
were stimulated with 16.5 ng/ml, 25 and 200 μg/ml of nicotine for
14 h, and the expression levels of CD80, CD86, CD40 and CD54 were
determined by flow cytometry. (A) Histographic presentation of CD80
expression on maDCs. (B) CD80 expression on maDCs (p=0.0256, imDC
control vs. Ni 16.5 ng/ml; p=0.0004, Ni 16.5 ng/ml vs. Ni 200
μg/ml). (C) Histographic presentation of CD86 expression on maDCs.
(D) CD86 expression on maDCs (p=0.0098, imDC control vs. Ni 16.5
ng/ml; p=0.0011, imDC control vs. Ni25 μg/ml; p=0.0437, imDC
control vs. Ni 200 μg/ml; p=0.0010, Ni 25 μg/ml vs. Ni 200 μg/ml).
(E) Histographic presentation of CD40 expression on maDCs. (F) CD40
expression on maDCs (p=0.0240, imDC control vs. Ni 16.5 ng/ml;
p=0.0459, imDC control vs. Ni 25 μg/ml; p<0.0001, imDC control
vs. Ni 200 μg/ml; p<0.0010, Ni 25 μg/ml vs. Ni 200 μg/ml). (G)
Histographic presentation of CD54 expression on maDCs. (H) CD54
expression on maDCs (p=0.0013, imDC control vs. Ni 16.5 ng/ml;
p<0.0001, Ni 16.5 ng/ml vs. Ni 25 μg/ml; p<0.0001, imDC
control vs. Ni 200 μg/ml). A representative flow cytometry analysis
out of 3 is shown; Student's t test. Ni, nicotine. |
The pro- or anti-apoptotic effects of
nicotine on imDCs were dose-dependent
As nicotine is thought to be toxic to cells, the
viabilities of imDCs stimulated with different doses of nicotine
were quantitated by flow cytometry using FITC-labeled Annexin V and
PI. The results showed that, 16.5–0.165 ng/ml of nicotine had no
effect on inducing DC apoptosis, but higher doses of nicotine
stimulation induced more than 90% cell apoptosis at the dose of
1.65 mg/ml and 165 μg/ml (Fig. 5B,
p<0.0001, imDC control vs. Ni 1.65 mg/ml; p<0.0001, imDC
control vs. Ni 165 μg/ml). In contrast to 1.65 mg/ml and 165 μg/ml
of nicotine, 16.5–0.165 ng/ml of nicotine treatment revealed an
anti-apoptotic effect on imDCs (Fig.
5B p=0.0046, imDC control vs. Ni 16.5 ng/ml; p=0.0405, imDC
control vs. Ni 0.165 ng/ml).
Discussion
In the past few years, a number of reports have
documented the biological effects of nicotine on DCs (4–9).
Aicher et al (5) reported
that nicotine dose-dependently enhanced DC co-stimulatory molecule
expression, enhanced IL-12 and IL-10 release, and augmented the T
cell priming ability of DCs. Our previous studies not only
characterized that nicotine has stimulatory effects on imDCs, but
also confirmed that nicotine-treated DCs exhibit anti-tumor effects
(6,7). In contrast, Nouri-Shirazi et
al (8) reported that, in the
presence of nicotine, monocyte-derived DCs manifested lower
endocytic and phagocytic activities, produced decreased levels of
proinflammatory cytokines, and had a reduced ability to stimulate
antigen-presenting cell-dependent T cell responses. Further studies
found that nicotine altered the biological activities of developing
mouse bone marrow-derived DCs (9).
Vassalo et al (13) found
that cigarette smoke extract (CSE), although not nicotine,
suppressed the DC-mediated priming of T cells in a mixed lymphocyte
reaction (MlR). Hence, there is a controversial conclusion
regarding the exact role of nicotine on DCs. Kawashima et al
reported that short-term exposure to nicotine enhanced lymphocyte
c-fos gene expression, but long-term exposure down-regulated nAchR
mRNA expression (15). In a fetal
thymus organ culture model, Middlebrook et al found that low
levels of nicotine (10−18–10−4M) increased
the number of immature T cells, but a higher dose
(>10−4 M) inhibited T cell development (16). The controversy regarding the
effects of nicotine on imDCs may be attributed to the differences
in experimental design, species, duration of exposure and
particularly the nicotine concentration used in these
experiments.
In the present study, we demonstrate that different
doses of nicotine have obviously different effects in inducing DC
apoptosis. High concentrations of nicotine (1.65 mg/ml and 165
μg/ml) were found to be toxic, leading to low cell viability
(Fig. 5). When 1.65 mg/ml of
nicotine was used to stimulate imDCs, nearly all cells were
undergoing apoptosis. There was no surprise to find that 200 μg/ml
of nicotine decreased the expression of CD80 and CD86 on imDCs
(Fig. 2) and supressed the
proliferation of DC-mediated T cells (8). Aicher et al and our previous
studies treated DCs with nicotine 16.5 ng/ml for 12 h, while
Nourii-Shirazi et al stimulated DCs with nicotine at a final
concentration of 200 μg/ml for 48 h, respectively, approximately
10,000-fold higher compared to the concentration of 16.5 ng/ml
(5–8). With 10 μg/ml of nicotine treatment,
Vassalo et al (13) also
acquired similar results of co-stimulatory molecule expression to
Aicher's data. Actually, Vassalo et al found that nicotine
as opposed to CSE, failed to inhibit DC-induced T cell priming, to
suppress the inflammatory up-regulation of co-stimulatory molecules
and the expression of chemotactic cytokine receptor 7 (CCR7) on
either imDCs or LPS-matured DCs (13). Since serum nicotine levels in
smokers are usually within the range of 10–100 ng/ml, never
exceeding the amount 100 μg/ml in vivo (17), the physiological relevance that
nicotine suppresses certain DC responses remains uncertain
(5).
Immunity requires DC maturation induced by microbial
endotoxins such as LPS, which increase the expression of
costimulatory molecules on the DC surface (18,19).
Our present study showed that lower doses of nicotine influenced DC
maturation and differentiation as revealed by the up-regulation of
costimulatory molecules CD80, CD40 and CD11c. In the presence of
LPS, in contrast to the 200 μg/ml of nicotine, 16.5 ng/ml of
nicotine stimulation obviously up-regulated the expression of
molecules CD80, CD86, CD40 and CD54 (Fig. 3). Consistent with our results,
Aicher et al reported that nicotine strongly activates
DC-mediated adaptive immunity (5).
They demonstrated that mouse bone marrow-derived competent DCs
exposed to 16.5 ng/ml of nicotine alone express higher levels of
MHCs and costimulatory molecules compared to the control DCs and
have a greater capacity to stimulate ovalbumin (OVA)-specific T
cell proliferation (5).
Conversely, Nouri-Shirazi et al reported that
nicotine-treated human DCs display an increased capacity for
antigen uptake, fail to fully up-regulate MHCs, hardly express CCR7
and display profoundly reduced Th1 promoting capacity (8). But, interestingly, their further
studies showed that while the presence of nicotine in the
microenvironment has no direct effect on competent mouse bone
marrow-derived DC function, it promotes the development of mouse
bone marrow-derived DC precursors into DCs with a semi-mature
phenotype revealed by higher expression of costimulatory molecules
CD80, CD86, CD40 and MHC II molecules and CCR7, and supports the
proliferation and differentiation of OVA-specific naïve T cells
into effector memory cells (9).
The differences in the DC preparations and treatments might account
for the discrepancies observed between ours and their studies. It
is worth mentioning that, when human monocyte-derived imDCs (day 6)
and murine bone marrow-derived imDCs (day 4) were used by
Nouri-Shirazi et al to study the effects of nicotine on
DC-mediated T cell priming, the conclusions were obviously
different. From our results, it appears that all of the factors of
the nicotinic microenvironment, nicotine doses used in the
experiment and DC maturation status, affect the functions of
DCs.
The data presented in this report offer new
information regarding the immunological alterations associated with
nicotine, particularly at the level of mouse DC differentiation.
This finding is important as it provides a rationale for further
investigation of the mechanisms by which nicotine influences DCs
in vivo and consequently hosts immunity using animal
models.
Acknowledgements
The authors would like to thank Professor X.T. Cao
(Second Military Medical University, Shanghai, China) and Y.H. Chen
(University of Pennsylvania, Philadelphia, USA) for kindly
providing the Hepa 1–6 cell lines and polishing the manuscript. In
addition, we thank Jin Hua Su and Fu Chen for their excellent
animal care. This study was supported by grants from the Natural
Science Foundation of Fujian Province of China (no. 2008J0112), the
Natural Science Foundation of Xiamen (no. 3502Z20104002) and the
Xiamen Science and Technology Key program (no. 3502Z20100006).
Abbreviations:
|
DCs
|
dendritic cells
|
|
imDCs
|
immature dendritic cells
|
|
maDCs
|
mature dendritic cells
|
|
nAChR
|
nicotinic acetylcholine receptor
|
|
Ni
|
nicotine
|
References
|
1
|
J AvilaJ Diaz-NidoTangling with
hypothermiaNat Med10460461200410.1038/nm0504-460
|
|
2
|
A MandavilliNicotine fixNat
Med10660661200410.1038/nm0704-660
|
|
3
|
C LibertInflammation – a nervous
connectionNature4213283292003
|
|
4
|
EK GuinetM YoshidaNouri-ShiraziNicotinic
environment affects the differentiation and functional maturation
of monocytes derived dendritic cells (DCs)Immunol
Lett954555200410.1016/j.imlet.2004.06.00315325797
|
|
5
|
AC AicherM HeeschenJP MohanptAM CookeAM
ZeiheS DimmelerNicotine strongly activates dendritic cell-mediated
adaptive immunity: potential role for progression of
atherosclerotic
lesionsCirculation107604611200310.1161/01.CIR.0000047279.42427.6D12566374
|
|
6
|
FG GaoDF WanJR GuEx vivo nicotine
stimulation augments the efficacy of therapeutic bone
marrow-derived dendritic cell vaccinationClin Cancer
Res1337063712200710.1158/1078-0432.CCR-07-002817575236
|
|
7
|
FG GaoHT LiZJ LiJR GuNicotine stimulated
dendritic cells could achieve anti-tumor effects in mouse lung and
liver cancerJ Clin
Immunol318088201110.1007/s10875-010-9459-520957418
|
|
8
|
M Nouri-ShiraziE GuinetEvidence for the
immunosuppressive role of nicotine on human dendritic cell
functionsImmunology109365373200310.1046/j.1365-2567.2003.01655.x12807482
|
|
9
|
M Nouri-ShiraziR TinajeroE GuinetNicotine
alters the biological activities of developing mouse bone
marrow-derived dendritic cells (DCs)Immunol
Lett109155164200710.1016/j.imlet.2007.02.00517368810
|
|
10
|
ST HannaNicotine effect on cardiovascular
system and ion channelsJ Cardiovasc
Pharmacol47348358200616633075
|
|
11
|
MH ZhangH TangZH GuoHZ AnXJ ZhuWG SongJ
GuoX HuangTY ChenJL WangXT CaoSplenic stroma drives mature
dendritic cells to differentiate into regulatory dendritic cellsNat
Immunol511241133200410.1038/ni113015475957
|
|
12
|
K ShortmanYJ LiuMouse and human dendritic
cell subtypesNat Rev Immunol2151161200210.1038/nri74611913066
|
|
13
|
R VassalloK TamadaJS LauPR KroeningL
ChenCigarette smoke extract suppresses human dendritic cell
function leading to preferential induction of Th-2 primingJ
Immunol17526842691200510.4049/jimmunol.175.4.268416081845
|
|
14
|
S BurgdorfA KautzV BöhnertPA KnolleC
KurtsDistinct pathways of antigen uptake and intracellular routing
in CD4 and CD8 T cell
activationScience316612616200710.1126/science.113797117463291
|
|
15
|
KawashimaKT FujiiThe lymphocytic
cholinergic system and its contribution to the regulation of immune
activityLife Sci74675696200310.1016/j.lfs.2003.09.03714654162
|
|
16
|
AJ MiddlebrookC MartinaY ChangRJ LukasD
DelucaEffects of nicotine exposure on T cell development in fetal
thymus organ culture: arrest of T cell maturationJ
Immunol16929152924200210.4049/jimmunol.169.6.291512218105
|
|
17
|
K MatsunagaTW KleinH FriedmanY
YamamotoInvolvement of nicotinic acetylcholine receptors in
suppression of antimicrobial activity and cytokine responses of
alveolar macrophages to Legionella pneumophila infection by
nicotineJ
Immunol16765186524200110.4049/jimmunol.167.11.651811714820
|
|
18
|
R AbeleR TampeThe ABCs of immunology:
structure and function of TAP, the transporter associated with
antigen processingPhysiology
(Bethesda)19216224200410.1152/physiol.00002.200415304636
|
|
19
|
AL AckermanP CresswellCellular mechanisms
governing cross-presentation of exogenous antigensNat
Immunol5678684200410.1038/ni108215224093
|
















