Introduction
During the development of acute pancreatitis (AP),
trypsinogen in pancreatic acinar cells is aberrantly activated by
external stimuli; the activated trypsin then activates additional
digestive zymogens, leading to the damage and self-digestion of
pancreatic acinar cells. Subsequently, during inflammatory cell
infiltration, a large number of inflammatory molecules enter the
circulatory system, causing systemic inflammatory response syndrome
(SIRS), multiple organ dysfunction syndrome (MODS) and even
death.
During this process, the abnormality of calcium
concentration and protein kinase, the reduction in adenosine
triphosphate (ATP) levels, the oxidative stress, as well as other
reactions in pancreatic acinar cells contribute to the development
of AP. All of these mechanisms are based on protein-kinase-mediated
intracellular signal transduction; thus, research on AP treatment
involving the targeting protein kinases may prove useful for the
treatment of multiple pathogeneses and may systematically inhibit
the development of AP.
In acute biliary pancreatitis, pancreatic enzyme
activation is caused by bile or some of its components, as well as
other causes. Bile acid directly acts on pancreatic acinar cells by
activating phosphatidylinositol 3-kinase (PI3K) to pathologically
increase the calcium level and activate digestive zymogens, cell
injury/death and inflammation pathways in cells (1). Bile acid also irritates inositol
1,4,5-trisphosphate (IP3) and ryanodine receptors, releasing the
calcium stored in the endoplasmic reticulum and zymogen granules of
acinar cells (2), thus leading to
pathological calcium transients in cells. In 2010, Perides et
al(3) demonstrated that
taurolithocholic acid 3-sulfate (TLC-S) causes an alteration in the
calcium concentration in cells by acting on the acinar cell surface
(Gpbar 1), and they concluded that biliary AP may be a
receptor-mediated disease.
The aim of this study was to provide evidence for
the development of AP treatment strategies targeting protein
kinases. Consequently, pancreatic acinar AR42J cells were treated
with TLC-S. Trypsinogen activation in pancreatic acinar cells was
observed, the differentially expressed protein kinase genes were
screened by gene chip analysis, and the functions of these kinases
were analyzed.
Materials and methods
Cell culture and processing
The AR42J rat pancreatic acinar cell line was
purchased from the China Center for Type Culture Collection (CCTCC;
Wuhan, China) and cultured in Ham’s F12 medium (F12K; Invitrogen,
Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS)
(HyClone, Logan, UT, USA), 100 U/ml penicillin and 100 μg/ml
streptomycin (Sigma, St. Louis, MO, USA). The cells were cultivated
in an incubator containing 5% CO2 at 37°C. The
activation of trypsinogen and the change in the proteome were
assessed 20 min following TLC-S (Sigma) treatment.
Assessment of trypsinogen activation
Acinar cells were cultured for 20 min in HBS-EDTA (5
mM HEPES, 0.15 M NaCl, 2 mM EDTA; pH 7.35) with 5 mM rhodamine 110,
bis-(CBZ-L-isoleucyl-L-prolyl-L-arginine amide) dihydrochloride
(BZiPAR; Molecular Probes®; Invitrogen), with the cell
concentration adjusted to 105 cells/ml. Trypsinogen
activation was then assessed using a flow cytometer (FACSDiva,
version 6.1; BD Biosciences, San Jose, NJ, USA) at an excitation
wavelength of 485 nm following treatment with 200 μM/l TLC-S for an
additional 20 min. The experiment was performed in triplicate.
Detection of gene expression using DNA
microarrays
The cells were collected, and total RNA was
extracted using TRIzol reagent (Invitrogen) according to the
manufacturer’s instructions. Gene expression analyses were
performed with the Rat 12×135K Gene Expression Array (NimbleGen
Systems, Inc., Madison, WI, USA). Preparation of cDNA from 5 μg of
total RNA, hybridizations, washes and detection were performed in
accordance to the NimbleGen gene expression analysis protocol
(NimbleGen Systems, Inc.); the slides were scanned using the Axon
GenePix 4000B microarray scanner.
Data analysis
Scanned images (TIFF format) were then imported into
NimbleScan software (version 2.5) for grid alignment and expression
data analysis. Expression data were normalized through quantile
normalization and the Robust Multichip Average (RMA) algorithm
included in the NimbleScan software. The Probe level
(*_norm_RMA.pair) files and Gene level (*_RMA.calls) files were
generated following normalization. All the gene level files were
imported into GeneSpring GX software (version 11.5.1; Agilent
Technologies, Inc., Santa Clara, CA, USA) for further analysis.
Differentially expressed genes were identified through fold change
filtering.
Based on the latest database of protein kinase
(http://kinasource.co.uk/Database/substrates.html),
differentially expressed kinase genes were selected. The
differentially expressed kinases genes which were singled out were
annotated in the Kyoto Encyclopedia of Genes and Genomes (KEGG;
http://www.kegg.jp) rat pathway. A visualized network
was drawn with the software cytoscape (version 2.6.3) to exhibit
the correlations between the kinases. Finally, each signaling
pathway that annotated the differentially expressed kinase genes
was imported into KEGG to search all related pathways, and the
correlation was exhibited with a visualized network using software
cytoscape (version 2.6.3).
Results
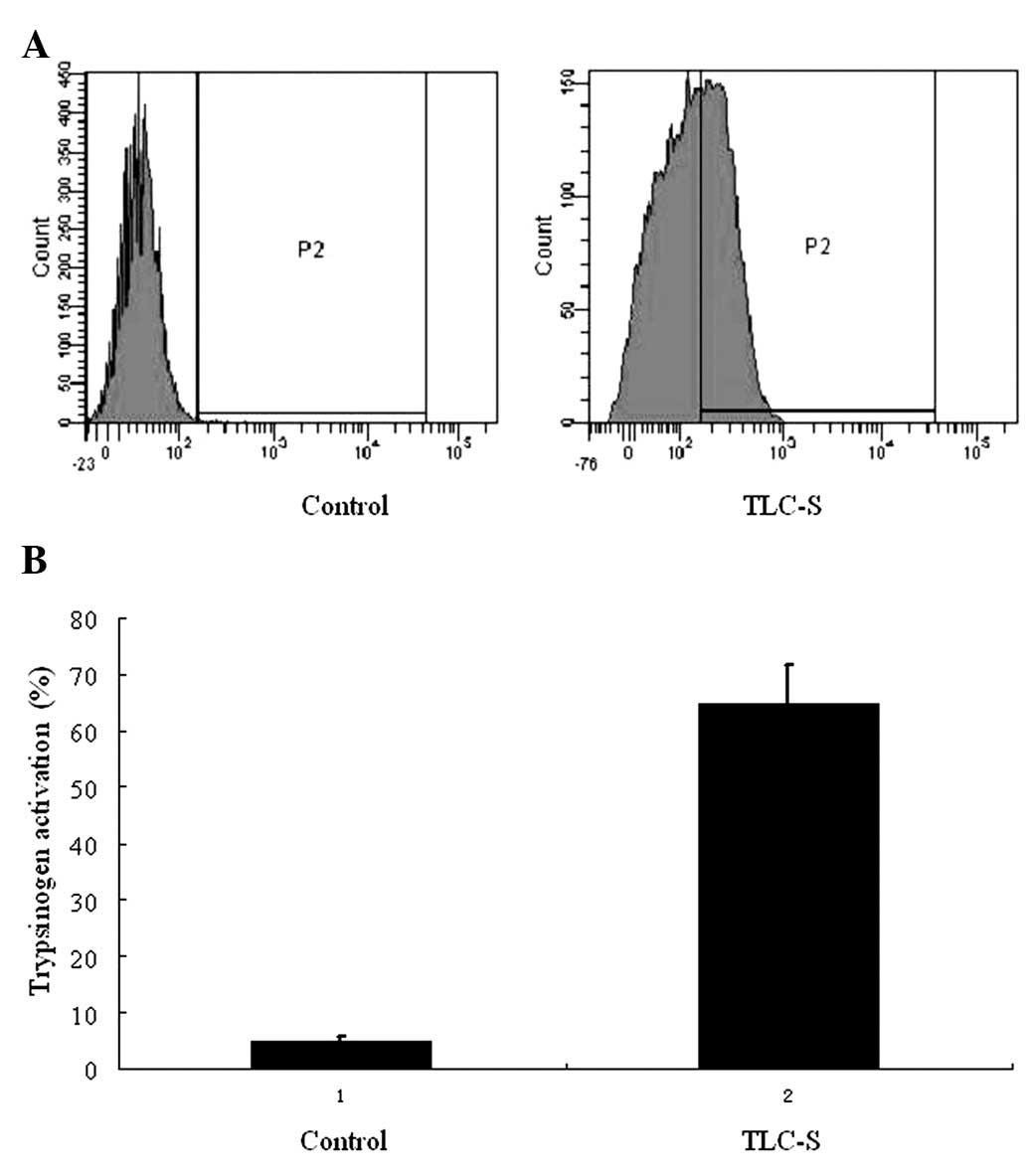
Trypsinogen activation
BZiPAR coupled with fluorescein rhodamine 110, can
not be excited to emit fluorescence prior to enzyme cleavage.
However, following trypsin enzymatic lysis, rhodamine 110 emits
green fluorescence excited by argon laser at 488 nm. As a result,
the degree of trypsinogen activation can be determined by detecting
the fluorescence intensity. In this experiment, there were only
trace amounts of activated trypsinogen found in the control group,
while a significantly higher level was detected by flow cytometry
following 20 min of treatment with TLC-S in the AR42J cells
(Fig. 1).
Identification of differentially
expressed protein kinase genes
A total of 11,292 genes were detected to be
expressed in the AR42J cells. In the TLC-S group, a total of 1,124
genes were upregulated and 498 were downregulated. Through
bioinformatics analysis, 22 differentially expressed protein kinase
genes were found in the 1,124 differentially expressed genes, among
which 19 genes were upregulated and 3 were downregulated (Table I).
 | Table IDifferently expressed protein kinase
genes in AR42J cells treated with taurolithocholic acid 3-sulfate
(TLC-S). |
Table I
Differently expressed protein kinase
genes in AR42J cells treated with taurolithocholic acid 3-sulfate
(TLC-S).
| Gene name | Gene ID | Description | Mode of
regulation | Fold change |
|---|
| Sgk196 | 306549 | Sugen kinase 196 | Upregulated | 2.0006287 |
| Map2k4 | 287398 | Mitogen-activated
protein kinase kinase 4 | Upregulated | 2.0351782 |
| Ptk2b | 50646 | Protein tyrosine
kinase 2 beta | Upregulated | 2.3132236 |
| Cdk1 | 54237 | Cyclin-dependent
kinase 1 | Upregulated | 4.0139747 |
| Lyn | 81515 | V-yes-1 Yamaguchi
sarcoma viral related oncogene homolog | Upregulated | 3.2749004 |
| Mapk14 | 81649 | Mitogen-activated
protein kinase 14 | Upregulated | 2.2141974 |
| Cdc42bpb | 113960 | CDC42 binding protein
kinase beta (DMPK-like) | Upregulated | 2.1441314 |
| Stk17b | 170904 | Serine/threonine
kinase 17b | Upregulated | 2.80085 |
| Tesk2 | 170908 | Testis-specific
kinase 2 | Upregulated | 2.557841 |
| Rps6ka2 | 117269 | Ribosomal protein S6
kinase, 90 kDa, polypeptide 2 | Upregulated | 3.3565648 |
| Sgk3 | 171498 | Serum/glucocorticoid
regulated kinase 3 | Upregulated | 2.0300796 |
| Ttk | 315852 | Tramtrack | Upregulated | 2.8380172 |
| Melk | 362510 | Maternal embryonic
leucine zipper kinase | Upregulated | 4.2185326 |
| Mast2 | 313819 |
Microtubule-associated serine/threonine
kinase 2 | Upregulated | 2.1736362 |
| Pak4 | 292756 | p21 protein
(Cdc42/Rac)-activated kinase 4 | Upregulated | 2.865645 |
| Bub1 | 296137 | Budding uninhibited
by benzimidazoles 1 homolog | Upregulated | 6.3756404 |
| Irak3 | 314870 | Interleukin-1
receptor-associated kinase 3 | Upregulated | 2.1035454 |
| Prkca | 24680 | Protein kinase C,
alpha | Upregulated | 2.1087615 |
| Mapk8 | 116554 | Mitogen-activated
protein kinase 8 | Upregulated | 2.7886798 |
| Fyn | 25150 | FYN oncogene
related to SRC, FGR, YES | Downregulated | −2.3253782 |
| Mst1 | 24566 | Macrophage
stimulating 1 | Downregulated | −3.9783108 |
| Ptk7 | 301242 | Protein tyrosine
kinase 7 | Downregulated | −3.8359256 |
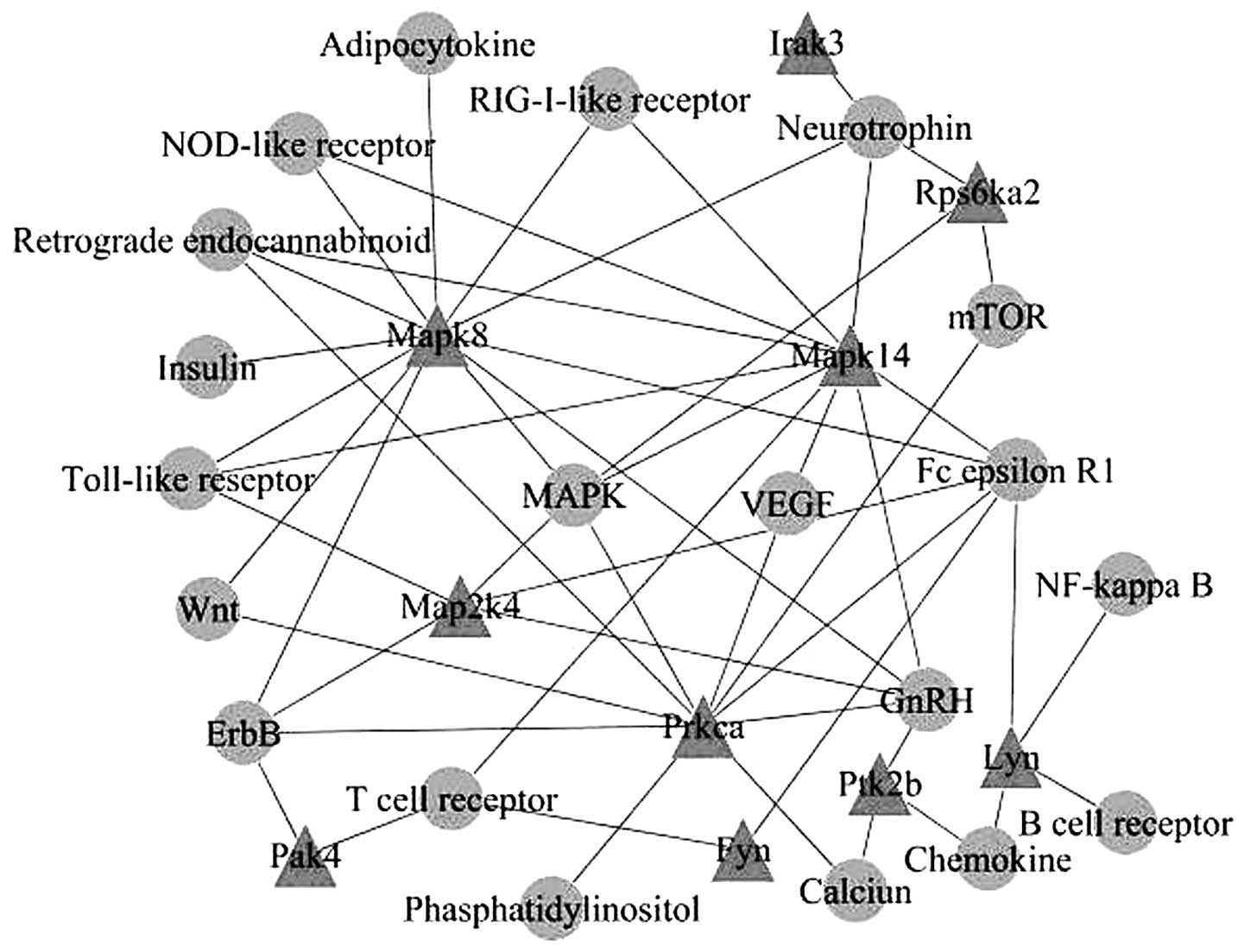
Based on the KEGG database, kinase genes of the same
KEGG pathways were connected to create a visualized network through
signaling pathways, and 10 nodes of kinases were identified, which
were mitogen-activated protein kinase (Mapk)8, Mapk14, Map2k4,
interleukin-1 receptor-associated kinase 3 (Irak3), ribosomal
protein S6 kinase, 90 kDa, polypeptide 2 (Rps6ka2), protein kinase
C, alpha (Prkca), v-yes-1 Yamaguchi sarcoma viral related oncogene
homolog (Lyn), protein tyrosine kinase 2 beta (Ptk2b), p21 protein
(Cdc42/Rac)-activated kinase 4 (Pak4) and FYN oncogene related to
SRC, FGR, YES (Fyn) (Fig. 2).
Subsequently, the interactions between the signaling
pathways were further analyzed and a network was created. MAPK and
calcium signaling pathways were found to be located at the center
of the network (Fig. 3).
Discussion
Trypsinogen activation in pancreatic acinar cells is
the initial factor involved in the development of all types of AP
and causes the self-digestion of cells. Shortly after the induction
of AP, immature secretory vesicles have been observed in the
cytoplasm, and the activity of trypsin has been detected in this
subcellular domain (4). However,
the underlying mechanisms of trypsinogen activation remain to be
elucidated. Several studies have shown that cathepsin and the
increased calcium concentration in cells are essential for the
activation of digestive enzymes (5). Over the past decade, the roles of
protein kinases and the signaling pathways associated with
trypsinogen activation have received increased attention (Table II).
| Table IIRegulation of trypsinogen activation
based on literature mining. |
Table II
Regulation of trypsinogen activation
based on literature mining.
| Regulators of
trypsinogen activation | Journal | Year of
publication |
Authors/(Refs.) |
|---|
| Cathepsins |
| Cathepsin B | J Clin Invest | 2010 | Reiser et
al(6) |
| Cathepsin B | Pancreatology | 2006 | Lindkvist et
al(7) |
| Cathepsin L |
Gastroenterology | 2010 | Wartmann et
al(8) |
| Calcium signaling
pathway |
| Calcineurin | Am J Physiol
Gastrointest Liver Physiol | 2009 | Shah et
al(9) |
| Calcium
release | Proc Natl Acad Sci
USA | 2009 | Gerasimenko et
al(10) |
| Calpain | Int J Exp
Pathol | 2009 | Weber et
al(11) |
| Ryanodine receptor
regulation calcium | Proc Natl Acad Sci
USA | 2005 | Husain et
al(12) |
| Protein kinase
signaling pathway |
| PKD/PKD1 | Am J Physiol
Gastrointest Liver Physiol | 2011 | Throwe et
al(13) |
| Rho-kinase | Br J Pharmacol | 2011 | Awla et
al(14) |
| MAPK | Am J Physiol
Gastrointest Liver Physiol | 2008 | Namkung et
al(15) |
| PKC | J Gastroenterol
Hepatol | 2008 | Gorelick et
al(16) |
| Pgk1, Pgk2 | Pancreatology | 2012 | Li et
al(17) |
| Endothelins |
| Endothelins | Exp Toxicol
Pathol | 2011 | Andrzejewska and
Dlugosz (18) |
| Endothelin-1
receptor antagonists | World J
Gastroenterol | 2005 | Andrzejewska et
al(19) |
| Enterokinases |
|
Enteropeptidase | Front Biosci | 2009 | Zheng et
al(20) |
| Enterokinase | Surgery | 2007 | Hartwig et
al(21) |
Protein kinase, also known as protein phosphakinase,
is a key regulator of numerous cellular functions. It changes the
molecular structure by adding a phosphate group to substrate
proteins in order to regulate protein activity, and it coordinates
cellular processes by acting on different signaling pathways.
Protein kinase is associated with almost every cellular function.
Protein kinase, which is important in regulating cell behavior, is
considered to be the target in the treatment for various diseases
including tumors, diabetes, osteoporosis, phlogosis and oculopathy
(22–24).
In the present study, the AR42J rat pancreatic
acinar cell line was used to establish a model of trypsinogen
activation in pancreatic cells following treatment with 200 μM
TLC-S (17). Total RNA from the
AR42J cells was then extracted to perform whole-genome expression
profile analysis, and 22 differentially expressed protein kinase
genes were identified. Ten nodes of protein kinase genes were
listed in a correlation diagram through analyzing KEGG pathways.
MAPK and calcium signaling pathways were found to play an important
role in trypsinogen activation.
The present study suggests that calcium signaling
pathways play an important role in trypsinogen activation, and that
Prkca and Ptk2b are associated with this signaling pathway. The
association between AP and [Ca2+]i in pancreatic acinar
cells has been previously investigated (9). In 1995, Ward et al(25) hypothesized that calcium overload in
acinar cells constitutes the main cause of AP development. Mooren
et al(26) identified an
increase in [Ca2+]i in acinar cells and an alteration of
the normal calcium signals. The early use of BAPTA-AM, a calcium
chelator, inhibits the above reaction. The abnormal release of
calcium in cells and the aberrant activation of zymogen in acinar
cells that cause self-digestion are known to be early events in the
progression of AP (27). According
to previous studies, the aberrant increase in [Ca2+]i in
acinar cells has been shown to directly trigger trypsinogen
activation and liquid cavity formation, since the treatment of
pancreatic acinar cells with large doses of cholecystokinin (CCK,
10 nmol/l) triggers trypsin activation of
[Ca2+]i-dependent top granules in acinar cells (28), and large doses of CCK cause changes
in the calcium dependence of acinar cells in cellular morphology
and the extensive replacement of zymogen granules by liquid cavity.
Raraty et al(29) showed
that the stably increased [Ca2+]i induces trypsinogen
activation. Shah et al(9)
observed that under the effect of FK506, a
calcium/calmodulin-protein-dependent phosphatase inhibitor, the
level of trypsinogen activation significantly decreased; a lower
level of blood amylase and the inflammatory factor, interleukin
(IL)-6, was also observed. The effect of FK506 was also
investigated by Ozawa (30).
Calcium/calmodulin calcineurin (PP2B) is controlled by many
factors; however, the main mechanisms are the aberrant increase of
[Ca2+]i in acinar cells and the alteration of calmodulin
concentration (31). PP2B has also
been shown to be the main target for the aberrant increase of
[Ca2+]i in pancreatic acinar cells. The aberrant
increase in [Ca2+]i in pancreatic acinar cells has been
shown to activate PP2B, which is considered to be an important step
in pathologic trypsinogen activation in pancreatic acinar
cells.
MAPK, one of the serine/threonine protein kinases,
presents in the majority of cells. It is one of the important
signaling systems where eukaryotic cells transduce extracellular
signals into cells to cause cellular reactions. Prior to
activation, MAPK is located in the cytoplasm, and it enters the
cell nucleus to activate the target gene following activation by
phosphorylation upon stimulation. MAPK affects the gene
transcription and regulation to influence the biological behavior
of cells, such as cell proliferation, differentiation,
transformation and apoptosis. The MAPK signaling pathway may be
induced by pro-inflammatory molecules and stress, and is involved
in most of the reactions of immunization and apoptosis caused by
pro-inflammatory molecules (32).
The present study identified the upregulated genes, Mapk14, Map2k4,
Mapk8, Rps6ka2 and Prkca, that are associated with the MAPK
signaling pathway, indicating that the MAPK signaling pathway plays
an important role in trypsinogen activation.
There are 3 subgroups in the MAPK family, p38, ERK
1/2 and JNK. p38 has been proven to be specifically associated with
the severity of AP and lung injury caused by severe AP. Thus,
injuries in the lungs and part of the pancreas are reduced by
inhibiting the activation of p38 (33). Certain studies have shown that
during the inflammatory processes, oxidative stress and
pro-inflammatory molecules are involved in the inflammatory cascade
process by enabling the signaling transduction pathway, and that
the activation of MAPK is of high importance during this process.
Pereda et al(34) also
demonstrated that the concurrent treatment of AP animal models with
oxypurinol, a p38 inhibitor, and pentoxifyllin, an ERK 1/2 and JNK
inhibitor, relieves inflammatory responses and reduces mortality.
Additionally, the inhibition of the p38, ERK1/2 and JNK signaling
pathways has been shown to have a less significant effect. The p38
signaling pathway specifically correlates with oxidative stress and
the ERK 1/2 and JNK pathways are clearly associated with the
expression of pro-inflammatory molecules. Since p38, ERK 1/2 and
JNK have an independent effect on the process of AP development,
concurrently inhibiting all 3 signaling pathways has a specific
effect on relieving AP symptoms. It has been reported that
oxidative stress causes MAPK activation and induces tumor necrosis
factor (TNF)-α (35). TNF-α has
also been suggested to be an initiator of cytokine cascade reaction
during AP development. Williard et al(36) showed that p38 is involved in the
activation of the pro-inflammatory nuclear transcription factor,
nuclear factor (NF)-κB, to upregulate the expression of
pro-inflammatory factors and aggregate AP. Samuel et
al(37) demonstrated that the
use of ERK 1/2 (PD98059), p38 (SB203580) and JNK (SP600125)
inhibitors to suppress their activation in a biliary AP model,
significantly reduced the expression of the pro-inflammatory
cytokine in pancreatic tissue. Liu et al(38) supported this finding by comparing
the serum and cytokine levels in the ascites of rats with severe
and mild AP.
In conclusion, protein kinases are important in AP
development, since they are associated with approximately every
mechanism of AP development. As a result, protein kinases
constitute potential drug targets for AP treatment. Gene
therapeutic methods, such as gene transfection and RNA interference
have proven to be successful in the treatment of AP in laboratory
experiments; however, their use in clinical practice is still
highly restricted. Targeting protein kinases as a form of therapy
should be investigated in clinical trials, since a number of kinase
inhibitors have been developed which may be used as a selective
target. Therefore, further studies on the role of protein kinases
in AP development are warranted, in order to develop a specific
kinase inhibitor for the target therapy of AP.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (30972907, 81070373). Financial support
was also provided from the Postdoctoral Science-Research
Developmental Foundation of Heilongjiang Province of China
(LBH-Q11038).
References
|
1
|
Voronina S, Longbottom R, Sutton R,
Petersen OH and Tepikin A: Bile acids induce calcium signals in
mose pancreatic acinar cells: implications for bile-induced
pancreatic pathology. J Physiol. 540:49–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gerasimenko JV, Flowerdew SE, Voronina SG,
et al: Bile acids induce Ca2+ release from both the
endoplasmic reticulum and acidic intracellular calcium stores
through activation of inositol trisphosphate receptors and
ryanodine receptors. J Biol Chem. 281:40154–40163. 2006.PubMed/NCBI
|
|
3
|
Perides G, Laukkarinen JM, Vassileva G and
Steer ML: Biliary acute pancreatitis in mice is mediated by the
G-protein-coupled cell surface bile acid receptor Gpbar1.
Gastroenterology. 138:715–725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thrower EC, Gorelick FS and Husain SZ:
Molecular and cellular mechanisms of pancreatic injury. Curr Opin
Gastroenterol. 26:484–489. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Halangk W and Lerch MM: Early events in
acute pancreatitis. Clin Lab Med. 25:1–15. 2005. View Article : Google Scholar
|
|
6
|
Reiser J, Adair B and Reinheckel T:
Specialized roles for cysteine cathepsins in health and disease. J
Clin Invest. 120:3421–3431. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lindkvist B, Fajardo I, Pejler G and
Borgström A: Cathepsin B activates human trypsinogen 1 but not
proelastase 2 or procarboxypeptidase B. Pancreatology. 6:224–231.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wartmann T, Mayerle J, Kähne T, et al:
Cathepsin L inactivates human trypsinogen, whereas cathepsin
L-deletion reduces the severity of pancreatitis in mice.
Gastroenterology. 138:726–737. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shah AU, Sarwar A, Orabi AI, et al:
Protease activation during in vivo pancreatitis is dependent on
calcineurin activation. Am J Physiol Gastrointest Liver Physiol.
297:G967–G973. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gerasimenko JV, Lur G, Sherwood MW, et al:
Pancreatic protease activation by alcohol metabolite depends on
Ca2+ release via acid store IP3 receptors. Proc Natl
Acad Sci USA. 106:10758–10763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weber H, Hühns S, Lüthen F and Jonas L:
Calpain-mediated breakdown of cytoskeletal proteins contributes to
cholecystokinin-induced damage of rat pancreatic acini. Int J Exp
Pathol. 90:387–399. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Husain SZ, Prasad P, Grant WM, Kolodecik
TR, Nathanson MH and Gorelick FS: The ryanodine receptor mediates
early zymogen activation in pancreatitis. Proc Natl Acad Sci USA.
102:14386–14391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thrower EC, Yuan J, Usmani A, et al: A
novel protein kinase D inhibitor attenuates early events of
experimental pancreatitis in isolated rat acini. Am J Physiol
Gastrointest Liver Physiol. 300:G120–G129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Awla D, Hartman H, Abdulla A, et al:
Rho-kinase signalling regulates trypsinogen activation and tissue
damage in severe acute pancreatitis. Br J Pharmacol. 162:648–658.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Namkung W, Yoon JS, Kim KH and Lee MG:
PAR2 exerts local protection against acute pancreatitis via
modulation of MAP kinase and MAP kinase phosphatase signaling. Am J
Physiol Gastrointest Liver Physiol. 295:G886–G894. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gorelick F, Pandol S and Thrower E:
Protein kinase C in the pancreatic acinar cell. J Gastroenterol
Hepatol. 23:S37–S41. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Z, Lu M, Chu J, Qiao X, Meng X, Sun B,
Zhang W and Xue D: Early proteome analysis of rat pancreatic acinar
AR42J cells treated with taurolithocholic acid 3-sulfate.
Pancreatology. 12:248–256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andrzejewska A and Dlugosz JW:
Differential effects of endothelins on histological and
ultrastructural changes and trypsinogen activation in the
secretagogue-induced acute pancreatitis in rats. Exp Toxicol
Pathol. 63:371–378. 2011. View Article : Google Scholar
|
|
19
|
Andrzejewska A, Dlugosz JW and
Augustynowicz A: Effect of endothelin-1 receptor antagonists on
histological and ultrastructural changes in the pancreas and
trypsinogen activation in the early course of caerulein-induced
acute pancreatitis in rats. World J Gastroenterol. 11:1115–1121.
2005. View Article : Google Scholar
|
|
20
|
Zheng XL, Kitamoto Y and Sadler JE:
Enteropeptidase, a type II transmembrane serine protease. Front
Biosci (Elite Ed). 1:242–249. 2009.PubMed/NCBI
|
|
21
|
Hartwig W, Kolvenbach M, Hackert T,
Fortunato F, Schneider L, Büchler MW and Werner J: Enterokinase
induces severe necrosis and rapid mortality in cerulein
pancreatitis: characterization of a novel noninvasive rat model of
necro-hemorrhagic pancreatitis. Surgery. 142:327–336. 2007.
View Article : Google Scholar
|
|
22
|
Morel M, Couturier J, Lafay-Chebassier C,
Paccalin M and Page G: PKR, the double stranded RNA-dependent
protein kinase as a critical target in Alzheimer’s disease. J Cell
Mol Med. 13:1476–1488. 2009.PubMed/NCBI
|
|
23
|
Kanda S, Miyata Y, Kanetake H and
Smithgall TE: Non-receptor protein-tyrosine kinases as molecular
targets for antiangiogenic therapy. Int J Mol Med. 20:113–121.
2007.PubMed/NCBI
|
|
24
|
Katoh Y and Katoh M: FGFR2-related
pathogenesis and FGFR2-targeted therapeutics (Review). Int J Mol
Med. 23:307–311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ward JB, Petersen OH, Jenkins SA and
Sutton R: Is an elevated concentration of acinar cytosolic free
ionized calcium the trigger for acute pancreatitis? Lancet.
346:1016–1019. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mooren FCh, Hlouschek V, Finkes T, et al:
Early changes in pancreatic acinar cell calcium signaling after
pancreatic duct obstruction. J Biol Chem. 278:9361–9369. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frick TW: The role of calcium in acute
pancreatitis. Surgery. 152:S157–S163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim JY, Kim KH, Lee JA, et al:
Transporter-mediated bile acid uptake causes
Ca2+-dependent cell death in rat pancreatic acinar
cells. Gastroenterology. 122:1941–1953. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Raraty M, Ward J, Erdemil G, Vaillant C,
Neoptolemos JP, Sutton R and Petersen OH: Calcium-dependent enzyme
activation and vacuole formation in the apical granular region of
pancreatic acinar cells. Proc Natl Acad Sci USA. 97:13126–13131.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ozawa T: FK506 induces biphasic
Ca2+ release from microsomal vesicles of rat pancreatic
acinar cells. Int J Mol Med. 18:187–191. 2006.PubMed/NCBI
|
|
31
|
Kessen U, Schaloske R, Aichem A and Mutzel
R: Ca(2+)/calmodulin-independent activation of calcineurin from
Dictyostelium by unsaturated long chain fatty acids. J Biol
Chem. 274:37821–37826. 1999.
|
|
32
|
Conze D, Krahl T, Kennedy N, et al: c-Jun
NH(2)-terminal kinase(JNK)1 and JNK2 have distinct roles in CD8(+)
T cell activation. J Exp Med. 195:811–823. 2002.
|
|
33
|
Yang J, Murphy C, Denham W, Botchkina G,
Tracey KJ and Norman J: Evidence of a central role for p38 map
kinase induction of tumor necrosis factor alpha in
pancreatitis-associated pulmonary injury. Surgery. 126:216–222.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pereda J, Sabater L, Cassinello N, et al:
Effect of simultaneous inhibition of TNF-alpha production and
xanthine oxidase in experimental acute pancreatitis: the role of
mitogen activated protein kinases. Ann Surg. 240:108–116. 2004.
View Article : Google Scholar
|
|
35
|
Araki Y, Andoh A, Yokono T, et al: The
free radical scavenger edaravone suppresses experimental closed
duodenal loop-induced acute pancreatitis in rats. Int J Mol Med.
12:121–124. 2003.PubMed/NCBI
|
|
36
|
Williard DE, Twait E, Yuan Z, Carter AB
and Samuel I: Nuclear factor kappa B-dependent gene transcription
in cholecystokinin and tumor necrosis factor-alpha-stimulated
isolated acinar cells is regulated by p38 mitogen-activated protein
kinase. Am J Surg. 200:283–290. 2010. View Article : Google Scholar
|
|
37
|
Samuel I, Zaheer S, Fisher RA and Zaheer
A: Cholinergic receptor induction and JNK activation in acute
pancreatitis. Am J Surg. 186:569–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu HS, Pan CE, Liu QG, Yang W and Liu XM:
Effect of NF-kappaB and p38 MAPK in activated monocytes/macrophages
on pro-inflammatory cytokines of rats with acute pancreatitis.
World J Gastroenterol. 9:2513–2518. 2003.PubMed/NCBI
|