Introduction
Alzheimer’s disease (AD) is one of the most common
neurodegenerative disorders morpho-pathologically characterized by
cellular Aβ amyloid plaque, intracellular neurofibrillary tangles
and extensive neuronal death. Besides these pathological
alterations, this disease is also associated with abnormalities in
the colinergic, serotoninergic, noradrenergic and dopaminergic
systems (1). These neurochemical
changes may be related to the abnormal blood glucose metabolism
that has been identified using positron emission tomography
(2). The reduction of glucose
utilization was not attributed to an insufficient supply of glucose
to the brain, but rather the decrease of glucose breakdown in brain
tissue (3,4). Furthermore, previous studies reported
that the activities of the number of enzymes and the expression of
the glucose transporter involved in glucose metabolism decreased in
the brain tissue of AD (5–7).
The existence of both insulin and insulin receptors
in the CNS is well established (8–9).
Insulin was first localized in the CNS of rats by
immunohistochemical staining (10). Subsequently, the insulin mRNA was
demonstrated to exist in various brain areas, suggesting that this
peptide is synthesized in the brain (11). In human brain, insulin regulates
enzymes associated with cerebral glucose metabolism via specific
high-affinity insulin receptors, which are different from
peripheral insulin receptors only in the amount of glycosylation
(12,13). Additionally, insulin binds to
insulin-like growth factor I (IGF-I) receptors and, via these
receptors, potentially exerting more generalized trophic effects on
neural cell and interacting with cholinergic neurotransmission
(14,15). The binding of insulin to its
receptor is followed by an autophosphorylation of tyrosine residues
at the β-chain of the insulin receptor resulting in a subsequent
activation of the intrinsic tyrosine kinase, which phosphorylates
the first known endogenous substrate insulin-receptor substrate-1
(IRS-1) (16). Phosphorylated
IRS-1 transfers its signal to a wide spectrum of cellular signal
transduction pathways (17). In
particular, AD patients exhibit alterations in insulin and IGF-1
levels and their receptors, leading to defective response to
insulin (18).
Furthermore, the study with transgenic (Tg) 2576
mice overexpressing the Swedish mutant human amyloid precursor
protein revealed an age-related cortical and hippocampal deposition
of β-amyloid plaques, as well as a decreased phosphofructokinase-C
(PFK-C) protein and mRNA level in cerebral cortical tissue.
Additionally, 24-month-old Tg2576 mice showed reduced enzyme
activity of PFK without affecting the mRNA levels of the other PFK
isoforms and fructose 1,6-bisphosphatase (FBPase) in comparison to
non-transgenic littermates (5).
However, no studies have been conducted thus far to investigate
whether the proteins involved in glucose metabolism are
significantly altered in neuron-specific enolase (NSE)/hPS2m Tg
mice that demonstrate AD-like pathology.
Therefore, the aim of this study was to investigate
whether AD pathology induced by overexpression of human mutant PS2
protein induces changes in glucose metabolism. The findings showed
that the enhancement of Aβ-42 peptides and γ-secretase activity in
NSE/hPS2m Tg mice significantly induced the defect of glucose
metabolism, including the decrease of insulin, the increase of
glucose, as well as the alteration of their related receptors and
the signal pathway. Furthermore, these results show that the
insulin treatment may decrease the level of Aβ-42 peptides in the
brain of NSE/hPS2m Tg mice.
Materials and methods
Care and use of NSE/hPS2m Tg mice
NSE/hPS2m Tg mice overexpressing human mutant PS2
(hPS2m, N141I), under the control of an NSE promoter were used in
this study (19,20) and were obtained from the Department
of Laboratory Animal Resources in Korea FAD. These Tg mice showed
behavioral dysfunction, Aβ-42 deposition and the induction of
caspase-3 and Cox-2 activities at 12 months of age. To analyze the
protein level and insulin concentration, a total of 10 mice were
used; five NSE/hPS2m Tg mice and five non-Tg littermates at 12
months of age. The mice were handled in a Pusan National
University-Laboratory Animal Resources Center accredited by the
Korea FDA in accordance with the USA NIH guidelines (accredited
unit no. 231). The mice were maintained in a specified
pathogen-free environment and were housed in cages under a strict
light cycle (light period for 12 h and dark period for 12 h) and
were given a standard irradiated chow diet (Purina Mills Inc.,
Milford, IN, USA) ad libitum.
Aβ-42 western blot analysis
For detection of the Aβ-42 level, the frozen brain
of mice was sectioned with scissors and homogenized in Pro-Prep™
Protein Extraction Solution [Intron Biotechechnology Co. Ltd.,
Seongnam, Korea, (50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM DTT, 0.5%
NP-40, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, proteinase
inhibitor)] with a glass homogenizer. The homogenate mixture was
centrifuged at 22,250 × g for 10 min at 4°C to eliminate the nuclei
and unbroken cells. The protein prepared from the brain was
separated by electrophoresis in a 4–20% SDS-PAGE gel for 3 h and
transferred to nitrocellulose membranes for 2 h at 40 V. Each
membrane was incubated separately with the primary anti-Aβ-42
antibody (MAB1560, 6E10; Chemicon International, Temecula, CA, USA)
overnight at 4°C. The membranes were washed with the washing buffer
(137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM
KH2PO4 and 0.05% Tween-20) and incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1,000
dilution; Zymed, South San Francisco, CA, USA) at room temperature
for 2 h. Blots were developed using a Chemiluminescence Reagent
Plus kit (ECL; Amersham Pharmacia, Piscataway, NJ, USA).
γ-secretase activity analysis
Detection of γ-secretase activity was performed
according to the manufacturer’s instructions (FP003; R&D System
Inc., Wiesbaden, Germany). Frozen brain tissues were sectioned with
scissors and homogenized in lysis solution and 1X cell extraction
buffer with a glass homogenizer into 0.5–2.0 mg/ml final
concentrations. The homogenate was separated by centrifugation at
22,250 × g for 15 min at 4°C and then supernatant was collected for
the protein and enzyme assays. Protein was assayed by the BCA
method (Pierce, Rockford, IL, USA) using an ELISA reader. For the
determination of γ-secretase activity, the 50 μl of tissue lysate
(25–200 μg) prepared from the brain extract was added to each well
in a 96-well microplate in triplicate and then 2X reaction buffer
(50 μl) was added. The substrate (5 μl) was added to each well and
the plate was incubated in the dark at 37°C for 1–2 h. Fluorescence
was measured at a wavelength of 335–355 nm using a Fluorescent
Microplate Reader FL600 (Bio-Tek Instrument, Inc., Winooski, VT,
USA).
Radioimmunoassay (RIA) and serum
biochemical analyses
Blood was collected from the abdominal vein of
NSE/hPS2m Tg mice and non-Tg littermates and then incubated at room
temperature for 30 min. The serum was separated by centrifugation
at 890 × g for 15 min at 4°C. Serum insulin concentration was
carried out as per the manufacturer’s instructions [Coat-A-Count
Insulin kit; Diagnostic Products Corp., LA, CA, USA) using a gamma
counter (Cobra 5010 Quantum, Cobra 5010 II; Packard Instrument Co.,
Inc., Meriden, CT, USA)]. The glucose concentration was assayed
according to the manufacturer’s instructions by Glucose Kit using
an Automatic Biochemical Analyzer (Hitachi 747, Tokyo, Japan).
Preparation of membrane protein
The brain was harvested from NSE/hPS2m and non-Tg
mice. Frozen brain was sectioned with scissors and homogenized in
buffer A [10 mM Tris (pH 7.4), 1 mM EDTA, 250 mM Sucrose,
proteinase inhibitor (pH 7.4)] with a glass homogenizer. The
homogenate mixture was centrifuged at 900 × g for 10 min at 4°C to
eliminate the unbroken cells. The supernatant was transferred to a
new tube and centrifuged at 110,000 × g for 75 min at 4°C to
collect the microsomal fraction. The pellet containing the
microsomal fraction was resuspended in lysis buffer A containing 1%
Triton X-100 for use in the western blot analysis.
Western blot analysis
The protein prepared from the brain tissues was
separated by electrophoresis in a 4–20% SDS-PAGE gel for 3 h and
transferred to nitrocellulose membranes for 2 h at 40 V. Each
membrane was incubated separately with the primary, anti-insulin
receptor α (sc-710; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA), anti-insulin receptor β (sc-711; Santa Cruz Biotechnology,
Inc.), anti-IGF receptor (I7151; Sigma-Aldrich, St. Louis, MO,
USA), anti-Glut-1 (sc-7903; Santa Cruz Biotechnology, Inc.),
anti-Glut-3 (sc7582; Santa Cruz Biotechnology, Inc.), anti-Glut-4
(400064; Calbiochem, Darmstadt, Hesse, Germany), anti-AKT (Ab3130;
Chemicon International), anti-p-AKT (Ab3132; Chemicon
International), anti-GSK (9332; Cell Signaling Technology, Inc.,
Boston, MA, USA), anti-p-GSK (9331; Cell Signaling Technology,
Inc.), anti-Tau (T7194; Sigma-Aldrich) and anti-p-Tau (T8069;
Sigma-Aldrich) antibodies overnight at 4°C. The membranes were
washed with washing buffer (137 mM NaCl, 2.7 mM KCl, 10 mM
Na2HPO4, 2 mM KH2PO4,
0.05% Tween-20) and incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG (1:1,000 dilution;
Zymed) at room temperature for 2 h. Blots were developed using a
Chemiluminescence Reagent Plus kit (ECL; Amersham Pharmacia).
Insulin treatments
Initially, the 12-month-old NSE/hPS2m Tg mice were
divided into three subgroups: No-treatment group [1X
phosphate-buffered saline (PBS)], low-dose group (0.2 units of
insulin), high-dose group (0.4 units of insulin). Insulin (Novolin
N, 100 IU/ml; GreenCross, Yongin, Korea) was subcutaneously
injected into NSE/hPS2m Tg mice in a volume of 0.2 ml dilution in
1X PBS solution for 4 weeks.
Statistical analysis
Tests for significance between non-Tg and Tg mice
were performed using a one-way ANOVA test of variance (SPSS for
Windows, Release 10.10, standard version; SPSS, Chicago, IL, USA).
Post-hoc tests of variance (SPSS for Windows, Release 10.10,
standard version) were used to determine significance between
insulin treatment and non-treatment Tg groups. Values were reported
as the mean ± standard deviation (SD). P<0.05 was considered to
indicate a statistically significant difference.
Results
Identification of AD phenotypes in
NSE/hPS2m Tg mice
To detect the phenotypes of AD resulting from the
overexpression of the hPS2m transgene, the Aβ-42 peptide and
activity of γ-secretase were quantified in total brain tissue of
12-month-old NSE/hPS2m Tg mice by western blot analysis and
γ-secretase activity assay, respectively. As shown in Fig. 1A, the level of Aβ-42 peptide was
significantly higher in the brain of the NSE/hPS2m Tg mice compared
with that of the non-Tg littermates. Also, γ-secretase activity was
significantly increased in the brain of the NSE/hPS2m Tg mice
(Fig. 1C). Furthermore, the level
of p-Tau (Thr 231 site) aggregated into the neurofibrillary tangle
in the cytoplasm of particular pyramidal neurons was significantly
increased in the brain of NSE/hPS2m Tg mice (Fig. 1B). These results suggest that
NSE/hPS2m Tg mice aged 12-months possessed the AD-like phenotypes
involving the deposition of Aβ-42, the increase of γ-secretase
activity and Tau hyperphosphorylation.
Alteration of insulin and glucose levels
in NSE/hPS2m Tg mice
To investigate whether cortical and hippocampal
disorganization caused by Aβ-42 peptides affected the insulin and
glucose levels, insulin and glucose concentrations were measured in
the serum obtained from NSE/hPS2m Tg mice. In the Coat-A-Count
Insulin kit analysis, the concentration of insulin was
significantly lower in the NSE/hPS2m Tg mice (2.1 mg/dl) than that
in the non-Tg littermates (6.2 mg/dl) (Fig. 1D). By contrast, the concentration
of glucose was significantly higher in the NSE/hPS2m Tg mice (210
mg/dl) compared with that in the non-Tg littermates (120 mg/dl)
(Fig. 1E). Therefore, changes of
the insulin and glucose concentration in Tg mice indicated that the
deposition of Aβ-42 peptide was directly associated with defects in
the glucose metabolism.
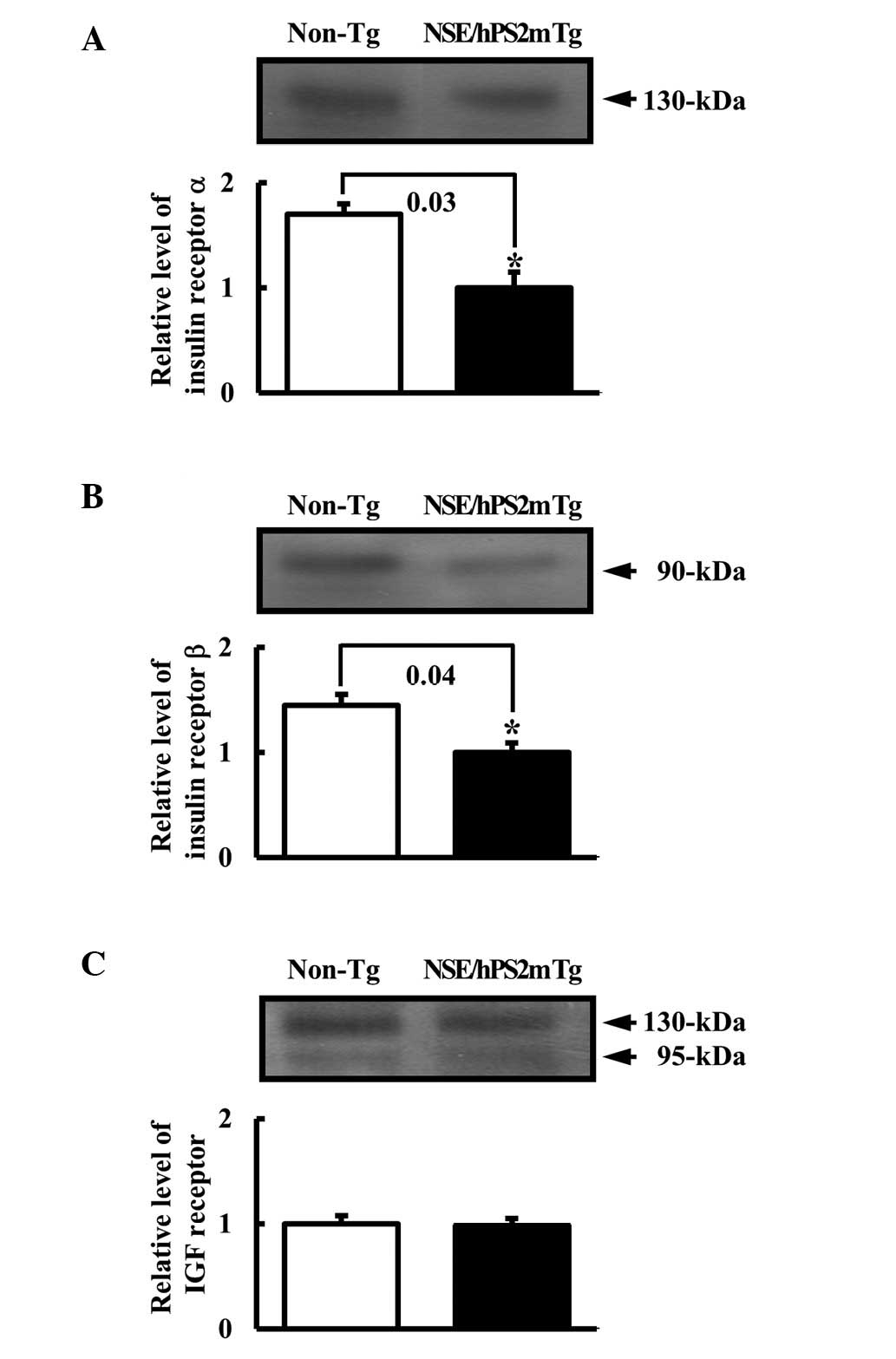
Differential expression of insulin
receptor and IFG receptor protein in NSE/hPS2m Tg mice
To determine whether the decrease in the insulin
concentration caused by Aβ-42 peptide affected its related receptor
expression, the expression levels of the insulin receptor and IGF
receptor protein were measured using western blot analysis from the
membrane protein of the brain tissues. Results of the western blot
analysis revealed that the expression of insulin receptors α and β
to be significantly reduced in the NSE/hPS2m Tg mice compared with
the non-Tg littermates (Fig. 2A and
B). However, these analyses also revealed no differences in the
expression level of IGF receptors in the brain between the
NSE/hPS2m Tg mice and non-Tg littermates (Fig. 2C). These results suggest that the
defect in glucose regulation was capable of significantly
decreasing the level of insulin receptors α and β, although the
level of IGF receptor was unaffected.
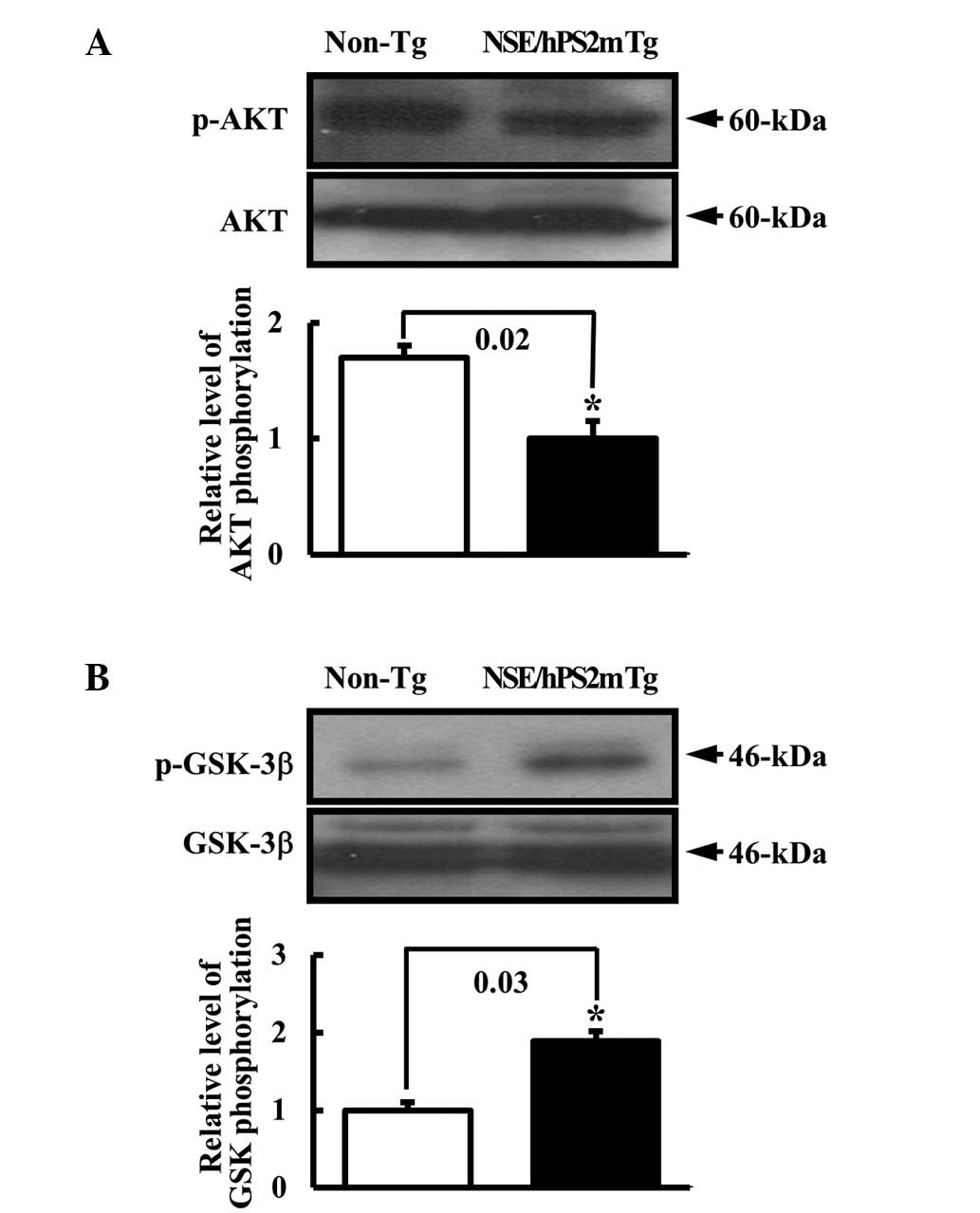
Effect of glucose regulation defect on
the insulin receptor signal transduction pathway
To test the hypothesis that the defect of the
glucose metabolism in the AD model would alter the phosphorylation
of signal protein in the insulin receptor signal pathway, the
phosphorylation rate of AKT and GSK proteins in the brain of
NSE/hPS2m Tg mice was detected. AKT2 is a key signaling molecule in
the insulin signaling pathway that induces glucose transport
(21). Glycogen synthase kinase 3
(GSK-3) involved in the Wnt signaling cascade may be inhibited
following phosphorylation by AKT (22). The level of p-AKT protein involved
in the downstream signal pathway of the insulin receptor was
reduced in the brain of NSE/hPS2m Tg mice compare with non-Tg
littermates (Fig. 3A). By
contrast, the p-GSK-3β level was higher in the NSE/hPS2m Tg mice
than the non-Tg littermates (Fig.
3B). Thus, this result suggests that the decrease in the
insulin receptors α and β significantly induced the change of
signal protein activation in the downstream signal pathway of the
insulin receptor, respectively.
Differential expression of glucose
transporter in NSE/hPS2m Tg mice
To determine whether the decrease of the glucose
level affected the expression of glucose transporter, western blot
analysis was performed to detect the expression level of the
glucose transporter in the membrane protein of mice brains.
Fig. 4 shows the differences in
the distribution of various molecular mass forms of Glut-1 and -3
in total cerebral homogenate. Two discrete bands at 45- and 55-kDa
are observed in total homogenate. The density of the 45- and 55-kDa
band in Glut-1 was significantly reduced in NSE/hPS2m Tg mice
compared with that of non-Tg littermates (Fig. 4A). The 45-kDa band in Glut-3 was
significantly reduced in the NSE/hPS2m Tg mice when this level was
compared with non-Tg littermates. However, the density of the
55-kDa band was not significantly different between the NSE/hPS2m
Tg and control mice (Fig. 4B). By
contrast, there was no difference in the Glut-4 protein level
between the NSE/hPS2m Tg mice and non-Tg littermates (Fig. 4C). These results suggest that the
defect of glucose regulation may be attributed to the decrease in
the expression level of Glut-1 and -3 proteins, but not for
Glut-4.
Effect of insulin administration on the
deposition of the Aβ-42 peptide
Previously, Boyt et al(23) suggested that glucose ingestion and
the subsequent elevation of plasma concentration of glucose and
insulin lead to a decrease in the level of amyloid precursor
protein in plasma. Therefore, to examine whether treatment of
insulin in the NSE/hPS2m Tg mice affected the concentration of the
Aβ-42 peptide, the insulin was injected into 12-month-old Tg mice.
In RIA and serum biochemical analysis, the insulin concentration
was significantly higher in the serum of insulin-treated mice than
that of non-treated Tg mice. However, the glucose concentration in
the insulin-treated mice was 3–4-fold lower than in the non-treated
Tg mice, respectively (Fig. 5A).
Furthermore, the western blot analysis revealed that the level of
Aβ-42 peptide in the brain of the insulin-treated mice was slightly
reduced compare with that of non-treated mice (Fig. 5B). Altering the level of Aβ-42
peptide from insulin-treated mice as shown by western blot
analysis, suggests the possibility that insulin treatment is
directly associated with a decrease in the Aβ-42 peptide (Fig. 5C).
Discussion
One of the key functional disturbances in AD is the
reduction in glucose utilization, which may be related to the
increased Aβ-42 peptide deposition that occurs in the neocortical
region and in walls of cerebral blood vessels. Glucose metabolism
is important in brain disorders, as glucose is the energy source in
the brain. In addition, the reduction in insulin concentration
could induce disturbing glucose metabolism (24–26).
Therefore, the present study was conducted with NSE/hPS2m Tg mice
and non-Tg mice to observe the correlation between the β-amyloid
peptide and glucose metabolism in the brain. A significant
behavioral dysfunction in the water maze test and the levels of
Aβ-42, caspase-3 and Cox-2 expression were especially observed in
the brains of NSE/hPS2m Tg mice at 12-months of age (19). These Tg mice showed a 40–50%
increase in the Aβ-42 peptide and γ-secretase activity from their
brains at 12-months of age (Fig. 1A
and B). A previous study reported that the NSE/hPS2m Tg mice
developed a greater number of fibrillar Aβ deposits in the cortex
and hippocampus than the non-Tg littermates (27). Furthermore, our results suggest
that the NSE/hPS2m Tg mice used in this study exhibit AD-like
phenotypes at 12-months of age.
Insulin is derived from a common precursor,
proinsulin, from which these peptides are released in equimolar
amounts by proteolytic cleavage (28). Insulin and Aβ peptide are also
common substrates for insulin-degrading enzyme (29), which is activated in various
tissues including brain tissues (30) and may be important in eliminating
toxic amyloidogenic peptides (31,32).
It has been demonstrated by cell culture and animal experiments
that insulin in the brain potently effects neuronal glucose
metabolism and cell differentiation (12,33).
In our study, the level of insulin and its receptor between
NSE/hPS2m Tg mice and non-Tg littermates were determined to examine
whether the incidence of AD was able to affect the glucose
metabolism pathway. RIA revealed that insulin concentration was
significantly reduced in the NSE/hPS2m Tg mice compared with the
non-transgenic mice. In addition, the expression of insulin
receptor α and β chain decreased in the brain of the NSE/hPS2m Tg
mice compared with that of the non-Tg littermates. With respect to
insulin receptors in NSE/hPS2m Tg mice, we have, to the best of our
knowledge, shown for the first time that the expression of insulin
receptors increased in the brain of NSE/hPS2m Tg mice thereby
increasing the Aβ-42 peptides by the overexpression of the mutant
PS2 gene under the control neuron-specific promoter. We have also
confirmed IGF-I receptor expression in the NSE/hPS2m Tg mice, as
shown earlier in AD patients (18,34,35).
The densities of these neurotrophic receptors were unchanged in
NSE/hPS2m Tg mice in contrast to the insulin receptor. These
results provide further evidence for specific involvement of brain
insulin receptors in the pathogenesis of AD.
Additionally, insulin binds to IGF receptors and,
via its receptor, possibly exerts more generalized trophic effects
on neural cells and interaction with cholinergic neurotransmission
(14,15). The binding of insulin to its
receptor is followed by the autophosphorylation of tyrosine
residues at the β-chain of insulin receptor resulting in subsequent
activation of the intrinsic tyrosine kinase, which phosphorylates
the initial endogenous substrate IRS-1 (16). Phosphorylated IRS-1 transfers
signals to a wide spectrum of cellular signal transduction pathways
(17). In the AD patients, the
activation of AKT was decreased in the signaling pathway of insulin
receptor downstream. By contrast, it was reported that the GSK-3β
and Tau protein on the downstream of AKT protein were significantly
activated in the brains of AD patients compared with the
age-matched controls (36). Our
results have shown that the phosphorylated AKT protein
significantly decreased in the brains of NSE/hPS2m Tg mice compared
with non-Tg littermates, while activated GSK-3 and Tau
significantly increased. These observations suggest that expression
of the hPS2m transgene might accelerate the pathogenic changes in
glucose metabolism defect, through an AKT, a GSK-3 and a Tau
phosphorylation, either directly or indirectly for underlying
AD.
Findings of previous studies have shown decreased
protein levels involved in glucose metabolism in AD patient brains.
In senile dementia of Alzheimer type, the concentration of plasma
glucose resulted in a significant increase compared with the
age-matched control group (37,38).
Significantly decreased cortical glucose transporter subtype
(Glut-1 and Glut-3) was identified in the brain of AD patients
compared with the age-matched controls (6,7,39).
Glut-1 and -3 are expressed the major glucose transporter in the
brain. Glut-1 can be detected as two molecular mass forms of 45-
and 55-kDa, which differ in their extent of glycosylation (40). Our data have identified reduced
Glut-1 and 3 proteins in the brain of NSE/hPS2m Tg mice, while no
difference was observed in the Glut-4 protein level. Furthermore,
the density of the 45- and 55-kDa bands in Glut-1 was significantly
reduced in NSE/hPS2m Tg mice compared with controls. The 55-kDa
band in Glut-3 was significantly reduced in the NSE/hPS2m Tg mice
compared to the non-Tg littermates. However, the density of the
45-kDa bands was not significantly different between the NSE/hPS2m
Tg mice and non-Tg littermates. These results suggest the defect of
glucose metabolism in the brain of NSE/hPS2m Tg mice is
significantly associated with the lower expression of the Glut-1
and -3 proteins, but not with Glut-4.
We also examined the effect of insulin on the
deposition of Aβ-42 peptides in the brain. Previous studies have
suggested that the subsequent elevation of plasma insulin leads to
a decrease in plasma amyloid precursor protein concentration
(23,41). In this study, when the NSE/hPS2m Tg
mice were treated with insulin by subcutaneous injection for 4
weeks, the level of the Aβ-42 peptide was slightly decreased in the
treated NSE/hPS2m Tg mice compared with the non-treated NSE/hPS2m
Tg mice. This observation suggests that insulin is important in
Aβ-42 peptide degradation processing, although the nature of this
role and the specific mechanisms remain to be elucidated.
Therefore, more studies are required to investigate the detailed
mechanism for the correlation between the insulin concentration and
amyloid precursor protein and the clinical significance of the
physiological changes in the insulin treatment condition.
Acknowledgements
We would like to thank Dr Jun Yong Cho at the Korea
National Sport University for consulting of glucose metabolism
analyses. This study was supported by the 2012 Specialization
Project Research Grant funded by the Pusan National University.
References
|
1
|
Gsell W, Moll G, Sofic E and Riederer P:
Cholinergic and monoaminergic neurotransmitter systems in patients
with Alzheimer’s disease and senile dementia of Alzheimer type: a
clinical evaluation. Dementias-neurochemistry, neuropathology,
neuroimaging, neuropsychology and genetics. Maurer K: Braunschweig;
Vieweg: pp. 25–51. 1993
|
|
2
|
Hoyer S: Senile dementia and Alzheimer’s
disease. Brain blood flow and metabolism. Prog Neuropsychopharmacol
Biol Psychiatry. 10:447–478. 1986.
|
|
3
|
Hoyer S, Oesterreich K and Wanger O:
Glucose metabolism as the site of the primary abnormality in
early-onset dementia of Alzheimer type? J Neurol. 235:143–148.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoyer S, Nitsch R and Oesterreich K:
Predominant abnormality in cerebral glucose utilization in
late-onset dementia of the Alzheimer type: a cross-sectional
comparison against advanced late-onset and incipient early-onset
cases. J Neural Transm Park Dis Dement Sect. 3:1–14. 1991.
View Article : Google Scholar
|
|
5
|
Bigl M, Apelt J, Eschrich K and Schliebs
R: Cortical glucose metabolism is altered in aged transgenic Tg2576
mice that demonstrate Alzheimer plaque pathology. J Neural Transm.
110:77–94. 2003.PubMed/NCBI
|
|
6
|
Mooradian AD, Chung HC and Shah GN: Glut-1
expression in the cerebral of patients with Alzheimer’s disease.
Neurobiol Aging. 18:469–474. 1997.
|
|
7
|
Simpson IA, Chundu KR, Davies-Hill T,
Honer WG and Davies P: Decreased concentrations of Glut1 and Glut3
glucose transporters in the brains of patients with Alzheimer’s
disease. Ann Neurol. 35:546–551. 1994.PubMed/NCBI
|
|
8
|
Baskin DG, Wilcox BJ, Figlewicz DP and
Dorsa DM: Insulin and insulin-like growth factors in the CNS.
Trends Neurosci. 11:107–111. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wozniak M, Rydzewski B, Baker SP and
Raizada MK: The cellular and physiological actions of insulin in
the central nervous system. Neurochem Int. 22:1–10. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harvankova J, Schmerchel D, Roth J and
Brownstein M: Identification of insulin in rat brain. Proc Natl
Acad Sci USA. 75:5737–5741. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Devasker SU, Giddings SJ, Rajakumar PA,
Carnaghi LR, Menon RK and Zahm DS: Insulin gene expression and
insulin synthesis in mammalian neuronal cells. J Biol Chem.
269:8445–8454. 1994.PubMed/NCBI
|
|
12
|
Hoyer S, Prem L, Sorbi S and Amsucci L:
Stimulation of glycolytic key enzymes in cerebral cortex by
insulin. Neuroreport. 4:991–993. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Pablo F and de la Rosa E: The
developing CNS: a scenario for the action of proinsulin, insulin
and insulin-like growth factors. Trends Neurosci. 18:143–150.
1995.PubMed/NCBI
|
|
14
|
Calissano P, Ciotti MT, Battistini L, Zona
C, Angelini A, Merlo D and Mercanti D: Recombination insulin-like
growth factor I exerts a trophic action and confers glutamate
sensitivity on glutamate-resistant cerebellar cells. Proc Natl Acad
Sci USA. 90:8752–8756. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Quirion R, Araujo DM, Lapehak PA, Seto D
and Chabot JG: Growth factors and lymphokines: modulators of
cholinergic neuronal activity. Can J Neurol Sci. 18:390–393.
1991.PubMed/NCBI
|
|
16
|
Sun XJ, Rothenberg P, Kahn CR, Backer JM,
Araki E, Wilden PA, Cahill DA, Goldstein BJ and White MF: Structure
of the insulin receptor substrate ISR-1 defines a unique signal
transduction protein. Nature. 352:73–77. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
White MF and Kahn CR: The insulin
signaling system. J Biol Chem. 269:1–4. 1994.
|
|
18
|
Frolich L, Blum-Degen D, Bernstein HG,
Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk
A, Hoyer S, Zochling R, Boissle KW, Jellinger K and Riederer P:
Brain insulin and insulin receptors in aging and sporadic
Alzheimer’s disease. J Neural Transm. 105:423–438. 1998.
|
|
19
|
Hwang DY, Chae KR, Kang TS, Hwang JH, Lim
CH, Kang HK, Goo JS, Lee MR, Lim HJ, Min SH, Cho JY, Hong JT, Song
CW, Paik SG, Cho JS and Kim YK: Alterations in behavior, amyloid
β-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of
Alzheimer’s disease. FASEB J. 16:805–813. 2002.
|
|
20
|
Hwang DY, Cho JS, Oh JH, Shim SB, Jee SW,
Lee SH, Seo SJ, Lee SK, Lee SH and Kim YK: Differentially expressed
genes in transgenic mice carrying human mutant presenilin-2
(N141I): correlation of selenoprotein M with Alzheimer’s disease.
Neurochem Res. 30:1009–1019. 2005.PubMed/NCBI
|
|
21
|
Garofalo RS, Orena SJ, Rafidi K, Torchia
AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks
JR, McNeish JD and Coleman KG: Severe diabetes, age-dependent loss
of adipose tissue, and mild growth deficiency in mice lacking
Akt2/PKB beta. J Clin Invest. 112:197–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meijer L, Flajolet M and Greengard P:
Pharmacological inhibitors of glycogen synthase kinase 3. Trends
Pharmacol Sci. 25:471–480. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boyt AA, Taddei TK, Hallmayer J,
Helmerhorst E, Gandy SE, Craft S and Martins RN: The effect of
insulin and glucose on the plasma concentration of Alzheimer’s
amyloid precursor protein. Neuroscience. 95:727–734. 2000.
|
|
24
|
Meier-Ruge WA and Bertoni-Freddari C:
Pathogenesis of decreased glucose turnover and oxidative
phosphorylation in ischemic and trauma-induced dementia of the
Alzheimer type. Ann N Y Acad Sci. 826:229–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hoyer S: Brain glucose and energy
metabolism abnormalities in sporadic Alzheimer disease. Causes and
consequences: an update. Exp Gerontol. 35:1363–1372. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hoyer S: The brain insulin signal
transduction system and sporadic (type II) Alzheimer disease: an
update. J Neural Transm. 109:341–360. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oyama F, Sawamura N, Kobayashi K,
Morishima-Kawashima M, Kuramochi T, Ito M, Tomita T, Maruyama K,
Saido TC, Iwatsubo T, Capell A, Walter J, Grunberg J, Ueyama Y,
Haass C and Ihara Y: Mutant presenilin 2 transgenic mouse: effect
on an age-dependent increase of amyloid beta-protein 42 in the
brain. J Neurochem. 71:313–322. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Polonsky KS and Rubenstein AH: C-peptide
as a measure of the secretion an hepatic extraction of insulin.
Pitfalls and limitations Diabetes. 33:486–494. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kurochkin IV and Goto S: Alzheimer’s
beta-amyloid peptide specifically interacts with and is degraded by
insulin degrading enzyme. FEBS Lett. 345:33–37. 1994.
|
|
30
|
Authier F, Posner BI and Bergeron JJ:
Insulin-degrading enzyme. Clin Invest Med. 19:149–160. 1996.
|
|
31
|
Perez A, Morelli L, Cresto JC and Castano
EM: Degradation of soluble amyloid beta-peptides 1–40, 1–42, and
the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer
disease and control brains. Neurochem Res. 25:247–255. 2000.
|
|
32
|
Vekrellis K, Ye Z, Qui WQ, Walsh D,
Hartley D, Cheseneau V, Rosner MR and Selkoe DJ: Neurons regulate
extracellular levels of amyloid-protein via proteolysis by
insulin-degrading enzyme. J Neurosci. 20:1657–1665. 2000.PubMed/NCBI
|
|
33
|
Henneberg N and Hoyer S: Short-term or
long-term intracerebroventricular (i. cv) infusion of insulin
exhibits a discrete anabolic effect on cerebral energy metabolism
in the rat. Neurosci Lett. 175:153–156. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
De Keyser J, Wilczak N and Goosens A:
Insulin-like growth factor-1 receptor densities in human frontal
cortex and white matter during aging, in Alzheimer’s disease, and
in Huntington’s disease. Neurosci Lett. 172:93–96. 1994.PubMed/NCBI
|
|
35
|
Crew FT, McElhaney R, Freund G, Ballinger
WE Jr and Raizada MK: Insulin-like growth factor I receptor binding
in brains of Alzheimer’s and alcoholic patients. J Neurochem.
58:1205–1210. 1992.
|
|
36
|
Stein TD and Johnson JA: Lack of
neurodegeneration in transgenic mice overexpressing mutant amyloid
precursor protein is associated with increased level of
transthyretin and the activation of cell survival pathways. J
Neurosci. 22:7380–7388. 2002.PubMed/NCBI
|
|
37
|
Craft S, Dagogo-Jack SE, Wiethop BV,
Murphy C, Nevins R, Fleschman S, Rice V, Newcomber JW and Cryer PE:
Effects of hyperglycemia on memory and hormone levels in dementia
of the Alzheimer type: a longitudinal study. Behav Neurosci.
107:926–940. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Manning CA, Ragozzino ME and Gold PE:
Glucose enhancement of memory in patients with probable senile
dementia of the Alzheimer’s type. Neurobiol Aging. 14:523–528.
1993.
|
|
39
|
Harr SD, Simonian NA and Hyman BT:
Functional alterations in Alzheimer’s disease: decreased glucose
transporter 3 immunoreactivity in the perforant pathway terminal
zone. J Neuropathol Exp Neurol. 54:38–41. 1995.
|
|
40
|
Sivitz WS, DeSautel PS and Pessin JE:
Regulation of the glucose transporter in developing rat brain.
Endocrinology. 124:1875–1880. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Craft S, Asthana S, Cook DG, Baker LD,
Cherrier M, Purganan K, Wait C, Petrova A, Latendresse S, Watson
GS, Newcomer JW, Schellenberg GD and Krohn AJ: Insulin
dose-response effects on memory and plasma amyloid precursor
protein in Alzheimer’s disease: interactions with apolipoprotein E
genotype. Psychoneuroendocrinology. 28:809–822. 2003.
|