Introduction
Primary gout affects 1–2% of adults (1–7) and
is one of the most common types of metabolic disease; a group of
diseases characterized by arthralgia, dysfunction, gouty
nephropathy and uremia. Although primary gout is a polygenic
hereditary disease, dietary habits and lifestyle changes, including
increased alcohol and excessive meat and seafood consumption, may
interact with the genetic factors to facilitate the pathogenesis
and development of gout. Additionally, insulin resistance,
abdominal obesity, dyslipidemia, arterial hypertension, diabetes
and metabolic syndrome are strongly associated with hyperuricemia
and gout (8,9). Previous studies have investigated the
candidate genes that control the production and clearance of uric
acid, that lead to hyperuricemia (2,10–12).
However, epidemiological evidence has demonstrated that <10% of
patients with hyperuricemia develop gout (13,14),
indicating that genes that are not linked to the metabolism of uric
acid may also contribute to the susceptibility for this
disease.
Gout is one of the most common types of
autoinflammatory arthritis and is characterized by an elevated
serum urate (uric acid) level and the intra-articular deposition of
monosodium urate (MSU) crystals. Uric acid and MSU are known to act
as host-derived danger-associated molecular patterns by directly
activating NALP3. The activation of NALP3 leads to the formation of
the inflammasome; a protein platform that mediates the processing
of intracellular interleukin (IL)-1β into its active form (15–17).
An association between NLRP3 gene (which encodes NALP3)
polymorphisms and autoinflammatory diseases, including type 1
diabetes and inflammatory bowel disease, has been suggested in
previous studies (18,19). Furthermore, multiple mutations in
the NLRP3 gene have been reported to be associated with
familial periodic fever syndromes, including Muckle-Wells syndrome
(MWS), familial cold-induced autoinflammatory syndrome (FCAS) and
neonatal-onset multisystem inflammatory disease (NOMID) (20–24).
It has been shown that serum IL-1β levels in these patients are
markedly increased (20–24). In each of these diseases, treatment
with the recombinant human IL-1 receptor antagonist (IL-1Ra)
anakinra induced the recovery of patients, which indicates that the
pathogenesis of these syndromes is strongly dependent on IL-1
(22). Gain-of-function mutations
of the NLRP3 gene were revealed in these patients, including
missense mutations in MWS, chronic infantile neurological cutaneous
and articular (CINCA) syndrome/NOMID and FCAS, which are located in
exon 3 of the NLRP3 gene (25). However, no previous studies have
investigated the association of polymorphisms and haplotypes in the
NLRP3 gene with susceptibility to primary gout.
The aim of the present study was to investigate the
assocation between genetic variants in 17 tagSNPs of the NLRP3 gene
and the susceptibility to primary gouty arthritis, via the
genotype-phenotype analysis of 480 primary gout and 480 control
patients.
Materials and methods
Study population
A total of 480 primary gout and 480 control patients
who were admitted to The Affiliated Hospital of Medical College,
Qingdao University (Qingdao, China) were enrolled in this study.
The gout patients were diagnosed by a clinical endocrinology
physician, according to the criteria set by the American Rheumatism
Association (26). Hyperuricemia
was defined as uric acid levels >420 mmol/l in males and
post-menopausal females, and >350 mmol/l in pre-menopausal
females. Informed consent was obtained from all the patients. The
clinical features of the patients were recorded. The criteria for
inclusion in the control group were that patients did not have a
family history of hyperuricemia and had no past medical history or
potential indications of hypertension, coronary atherosclerotic
heart disease, diabetes, hyperlipidemia, cancer, hepatic disease or
renal disease.
The study protocol was in accordance with the Ethics
Guidelines of the 1975 Declaration of Helsinki and was approved by
The Affiliated Hospital of Medical College, Qingdao University.
Measurement of biochemical
parameters
The plasma levels of blood glucose, creatinine, uric
acid, total cholesterol (TC) and triglycerides (TG) in all the
patients were measured using an automated multichannel chemistry
analyzer (Toshiba 200FR; Toshiba Medical Systems, Tokyo,
Japan).
Genotype analysis
Peripheral blood samples were obtained from the
patients and DNA samples were preserved at −20°C. Genomic DNA was
isolated from the peripheral blood leukocytes and extracted
according to the manufacturer’s instructions (QIAamp DNA Blood Mini
kit; Qiagen, Hiden, Germany). The ODs at 260 and 280 nm were
determined using a UV spectrophotometer (DU-650; Beckman Coulter,
Inc., Miami, FL, USA) to determine the DNA concentration.
Potentially functional SNPs of the NLRP3 gene
were selected from the HapMAP database (http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap24_B36/).
Seventeen tagSNPs were selected based on the standard
(r2, ≥0.08; MAF, >0.05). SNP genotyping was performed
using the Sequenom MassARRAY iPLEX platform (Table II). The full length of the
NLRP3 gene is ~30 kbp, including 9 exons and 8 introns. A
total of 12 pairs of primers were used for the polymerase chain
reaction (PCR) and these were designed using the GeneTools and
DNAMAN softwares, according to the manufacturer’s instructions. PCR
was carried out in a reaction volume of 20 μl, containing 50 ng of
genomic DNA, 200 μM dNTP, 2.5 units of Taq DNA polymerase
(Promega Corporation, Madison, WI, USA) and 200 μM of primers. The
conditions of the PCR were as follows: 94°C for 2 min, 35 cycles of
94°C for 30 sec, an annealing temperature reduced to 64°C for 30
sec and 72°C for 1 min. The PCR products were analyzed using
electrophoresis on 1.0% agarose gel. For quality control,
genotyping was performed without knowledge of the case/control
status of the subjects and 5% of the total number of case and
control patients were selected at random and re-genotyped by
different investigators; the reproducibility was 100%.
 | Table IIGenotype distribution and allele
frequencies of the NLRP3 gene in the cases and control
patients. |
Table II
Genotype distribution and allele
frequencies of the NLRP3 gene in the cases and control
patients.
| SNP | Position | Genotype
frequency | P-value | P-value for the HWE
in controls | Alleles | MAFs | MAFs |
|---|
|
|---|
| Cases | Controls |
|---|
|
|
|
|---|
| 1/1 | 1/2 | 2/2 | 1/1 | 1/2 | 2/2 | Cases | Controls |
|---|
| rs4925648 | 247580568 | 319 | 146 | 14 | 320 | 127 | 25 | 0.112 | 0.81 | C/T | 0.14/T | 0.182 | 0.188 |
| rs7512998 | 247583221 | 387 | 87 | 5 | 393 | 79 | 2 | <0.05 | 0.59 | C/T | 0.136/C | 0.101 | 0.088 |
| rs4925650 | 247584075 | 153 | 237 | 90 | 158 | 209 | 105 | 0.23 | 0.91 | A/G | 0.357/A | 0.434 | 0.444 |
| rs12137901 | 247584591 | 281 | 164 | 29 | 272 | 145 | 46 | 0.08 | 0.41 | C/T | 0.285/C | 0.234 | 0.256 |
| rs10754555 | 247584643 | 205 | 212 | 60 | 214 | 183 | 73 | 0.17 | 0.90 | C/G | 0.418/G | 0.348 | 0.350 |
| rs3806266 | 247587065 | 410 | 66 | 3 | 408 | 63 | 2 | 0.89 | 0.92 | A/G | 0.025/A | 0.075 | 0.071 |
| rs7525979 | 247587408 | 294 | 167 | 19 | 301 | 149 | 22 | 0.53 | 0.65 | C/T | 0.122/T | 0.214 | 0.204 |
| rs3806268 | 247587477 | 132 | 247 | 101 | 141 | 218 | 113 | 0.26 | 0.96 | A/G | 0.41/A | 0.532 | 0.530 |
| rs4925651 | 247587531 | 479 | 1 | 0 | 469 | 3 | 0 | 0.31 | 0.95 | G/T | 0.053/T | 0.001 | 0.003 |
| rs3738448 | 247593142 | 291 | 170 | 19 | 297 | 152 | 23 | 0.50 | 0.59 | A/C | 0.075/C | 0.783 | 0.790 |
| rs10925019 | 247595850 | 266 | 185 | 29 | 268 | 171 | 33 | 0.68 | 0.91 | C/T | 0.169/T | 0.253 | 0.251 |
| rs4612666 | 247599070 | 142 | 247 | 91 | 149 | 229 | 96 | 0.62 | 0.66 | C/T | 0.387/T | 0.447 | 0.444 |
| rs10754557 | 247599232 | 245 | 196 | 38 | 242 | 185 | 47 | 0.53 | 0.87 | A/G | 0.484/G | 0.284 | 0.294 |
| rs12143966 | 247601357 | 132 | 239 | 108 | 132 | 231 | 106 | 0.98 | 0.99 | A/G | 0.377/A | 0.475 | 0.472 |
| rs12239046 | 247604258 | 142 | 246 | 91 | 148 | 229 | 95 | 0.68 | 0.69 | C/T | 0.402/T | 0.447 | 0.444 |
| rs10159239 | 247607642 | 154 | 229 | 97 | 134 | 230 | 109 | 0.36 | 0.28 | A/G | 0.417/G | 0.441 | 0.474 |
| rs12565738 | 247609328 | 401 | 74 | 4 | 413 | 61 | 0 | 0.06 | 0.20 | C/T | 0.167/T | 0.086 | 0.064 |
Statistical analysis
Stata 8.0 (StataCorp, College Station, TX, USA) was
used to perform statistical analysis. Continuous variables are
expressed as the mean ± standard deviation (SD), while categorical
variables are expressed as frequencies and percentages. The
χ2 test and Student’s t-test were used to compare
clinical characteristics between the two groups. The χ2
test was also used to determine the Hardy-Weinberg equilibrium
(HWE) in the control patients. Differences between the
non-continuous variables, genotype distribution and allele
frequency were assessed using the χ2 test. Haploview 4.2
was used to calculate linkage disequilibrium blocks and haplotype
association risk (27). The odds
ratio (OR) and 95% confidence interval (CI) were evaluated using
binary logistic regression analysis. Comparisons were two-sided and
P<0.05 was was considered to indicate a statistically
significant difference.
Results
Subject characteristics
The clinical characteristics of the subjects are
provided in Table I. The gout
patients had significantly higher blood uric acid, triglyceride and
blood glucose levels, body mass index and waist-hip ratio compared
with the control patients (P<0.01). No significant difference
was observed in serum cholesterol, urea nitrogen and creatinine
levels between the two groups (Table
I).
| Table IDemographic and clinical
characteristics of the study population (mean ± SD). |
Table I
Demographic and clinical
characteristics of the study population (mean ± SD).
| Characteristics | Gout patients
(n=480) | Control patients
(n=480) | P-value |
|---|
| Age (years) | 52.48±13.12 | 60.57±9.45 | <0.001a |
| Height (cm) | 173.37±5.23 | 169.43±6.95 | <0.001a |
| Weight (kg) | 80.92±12.12 | 71.21±9.65 | <0.001a |
| BMI
(kg/m2) | 26.87±3.46 | 24.78±2.90 | <0.001a |
| Circumference of
waist (cm) | 96.41± 8.89 | 91.60± 7.06 | <0.001a |
| Circumference of
hip (cm) | 104.59±7.33 | 101.11±6.14 | <0.001a |
| W/H ratio | 0.92±0.52 | 0.91±0.51 | <0.001a |
| Systolic blood
pressure (mmHg) | 136.75±19.84 | 132.48±16.49 | <0.001a |
| Diastolic blood
pressure (mmHg) | 87.84±12.16 | 84.70±9.76 | <0.001a |
| Blood glucose
(mmol/l) | 6.16±1.84 | 5.64±1.22 | <0.001a |
| TG (mmol/l) | 2.40±2.23 | 1.48±0.98 | <0.001a |
| TC (mmol/l) | 5.26±1.30 | 5.27±0.84 | 0.846 |
| Urea nitrogen
(mmol/l) | 6.12±2.72 | 5.87±1.42 | 0.073 |
| Creatinine
(μmol/l) | 91.45±33.53 | 94.17±15.93 | 0.110 |
| Uric acid
(μmol/l) | 495.98±133.08 | 322.94±52.90 | <0.001a |
HWE and linkage disequilibrium
(LD)/haplotype analysis
The genetic frequency of rs7512998 was significantly
different between the gout and control patients (P<0.05;
Table II), whereas no significant
differences were observed for the remaining SNPs. Seventeen SNPs
conformed to the HWE in the control patients (P>0.05; Table II). The minor allele frequencies
of SNPs containing rs4925651, rs3806266 and rs3738448 were notably
low (<10%).
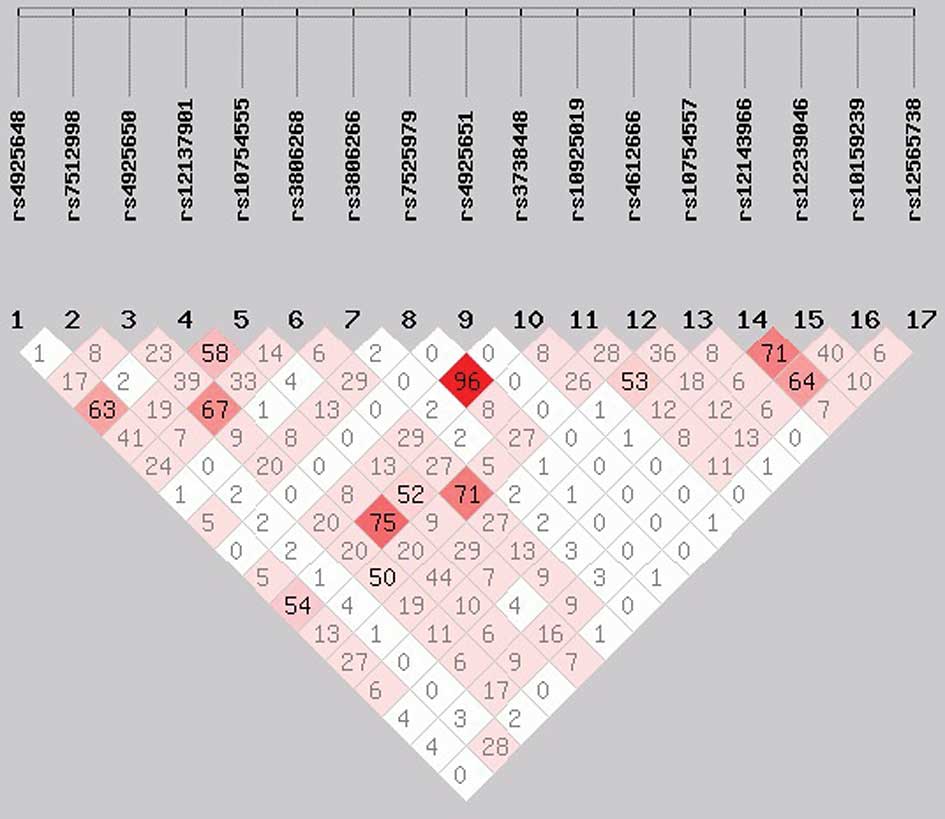
To investigate the haplotype association among the
17 SNPs of the NLRP3 gene, the r2 measure of LD
was estimated and demonstrated that only one polymorphism,
rs7525979, had a high LD (r2>0.9; Figs. 1 and 2). No individual SNP was demonstrated to
be significantly associated with primary gouty arthritis.
Among 134 haplotypes in the 17 SNPs, 8 haplotypes of
the NLRP3 gene accounted for 98% of the subjects and were
included in the analysis (Table
III). The haplotype CTATCAGCGCCCAGTGC was the most common among
the case and control groups, with a frequency of 0.224 and 0.243,
respectively. However, the ORs of the 8 haplotypes were not
identified to be significantly associated with gouty arthritis
(P>0.05 for all the 8 haplotypes).
| Table IIIAssociation between 8 haplotypes of
the NLRP3 gene and the risk of gouty arthritis. |
Table III
Association between 8 haplotypes of
the NLRP3 gene and the risk of gouty arthritis.
| Haplotype | Cases (freq) | Controls
(freq) | χ2 | P-value | Odds ratio (95%
CI) |
|---|
|
CCGTGAGCGCCCAGTAT | 44.96 (0.048) | 36.93 (0.041) | 0.5 | 0.475374 | 1.178
(0.751–1.846) |
|
CTATCAACGCCCAGCGC | 45.98 (0.049) | 38.77 (0.043) | 0.4 | 0.54526 | 1.146
(0.737–1.783) |
|
CTATCAGCGCCCAACAC | 83.23 (0.088) | 74.51 (0.083) | 0.2 | 0.651918 | 1.080
(0.774–1.507) |
|
CTATCAGCGCCCAGTGC | 211.20 (0.224) | 216.97 (0.243) | 0.8 | 0.375724 | 0.901
(0.715–1.135) |
|
CTGCGGGCGCTTGACAC | 43.71 (0.046) | 47.99 (0.054) | 0.5 | 0.497148 | 0.863
(0.564–1.320) |
|
CTGTCGGTGACTAACAC | 105.93 (0.112) | 93.73 (0.105) | 0.4 | 0.543856 | 1.098
(0.812–1.485) |
|
CTGTCGGTGACTAGTGC | 44.57 (0.047) | 38.92 (0.044) | 0.2 | 0.663469 | 1.104
(0.708–1.721) |
|
TTGCGGGCGCTTGACAC | 87.18 (0.092) | 90.73 (0.101) | 0.4 | 0.550771 | 0.908
(0.662–1.246) |
Polymorphisms of the NLRP3 gene and
primary gouty arthritis
Conditional logistic regression analysis
demonstrated that the 17 SNPs were not significantly different
between the groups and no significant ORs for the risk of gouty
arthritis were noted (data not shown).
Discussion
To the best of our knowledge, the present study is
the first investigation on the potential association of the risk of
gouty arthritis with polymorphisms and haplotypes in a presumptive
promoter region of the NLRP3 gene. However, this study did
not demonstrate a significant association between SNPs of the
NLRP3 gene and gouty arthritis. Female gout patients were
not included in the present study, since the incidence of gouty
arthritis in females is lower compared with that in males.
A previous genome-wide association study
demonstrated that the NLRP3 inflammasome gene is associated with a
number of autoimmune diseases, including familial cold urticaria
(28) and MWS (29), and numerous inflammatory diseases
(30), including Crohn’s disease
(31), obesity-induced
inflammation and insulin-resistant FCAS (32). The activation of NLRP3
inflammasomes results in an inflammatory response driven by the
secretion of IL-1β, a pro-fibrotic cytokine, which is essential in
the pathogenesis of inflammation-induced pulmonary fibrosis
(33). Furthermore, NLRP3
inflammasomes induce the exposure of MHC-II on
macrophage/antigen-presenting cell surfaces for a rapid non-self
antigen presentation (34) and are
important in the maturation and activation of dendritic cells
(35,36). Thus, the activation of NLRP3
drives local inflammation and the acquired immune response. More
than 30 different SNPs located within exon 3 of the NLRP3
gene that encode the nucleotide binding site domain and boundary
regions have been identified (32). However, results of the present
study have shown that mutations of the NLRP3 gene were not
associated with the risk of gouty arthritis. There are several
possible explanations for this result: The full-length of the
NLRP3 gene is ~30 kbp, including 9 exons and 8 introns; in
the present study, only 17 SNPs were selected from 388 SNPs of the
NLRP3 gene, which may not account for all the potentially
susceptible SNPs. Furthermore, the sample size in this study was
small and the statistical power to determine the authentic
susceptible SNP loci in low allele frequencies was low. Therefore,
further studies are required to confirm the results of the present
study.
There were a number of limitations in the present
study. Firstly, we focused solely on genetic variants and only 17
SNPs were selected among 388 SNPs of the NLRP3 gene.
Therefore, more functional studies on the effects of genetic
variants on transcription factor binding activity and changes at
the protein level are required. Secondly, although the association
between polymorphisms in the NLRP3 gene and the risk of
gouty arthritis was investigated, the potential association between
additional risk factors and gouty arthritis were not assessed.
Therefore, a more detailed data analysis may allow the association
between additional risk factors and genetic variants to be
elucidated. Thirdly, the sample size of the present study was small
and all the subjects were of the same ethnicity; thus, our results
may not be representative of the various ethnicities in China and
other countries. The experiments performed in this study should be
repeated in other independent populations using larger sample
sizes. Moreover, the gout phenotypes were assessed using the
medical history of the patients. The data for several phenotypic
disease characteristics, including the duration of gout history,
disease location and whether the patient had metabolic syndrome,
insulin resistance, cardiovascular disease or chronic syndrome,
were not available. Therefore, further studies should consider the
detailed clinical histories of the patients.
To the best of our knowledge, this is the first
study to investigate the association between SNPs of the
NLRP3 gene and the risk of primary gouty arthritis; however,
no association was detected. Clinical studies and functional
analysis are required to investigate the mechanisms underlying the
association between NLRP3 gene polymorphisms and/or
mutations and the risk of primary gouty arthritis.
Acknowledgements
This study was supported by a grant from the Qingdao
Municipal Science and Technology Commission (project no.
10-3-4-7-2-jch to Dong-Mei Meng).
References
|
1
|
Roddy E, Zhang W and Doherty M: The
changing epidemiology of gout. Nat Clin Pract Rheumatol. 3:443–449.
2007. View Article : Google Scholar
|
|
2
|
Terkeltaub R: Gout in 2006: the perfect
storm. Bull NYU Hosp Jt Dis. 64:82–86. 2006.PubMed/NCBI
|
|
3
|
Falasca GF: Metabolic diseases: gout. Clin
Dermatol. 24:498–508. 2006. View Article : Google Scholar
|
|
4
|
Zaka R and Williams CJ: New developments
in the epidemiology and genetics of gout. Curr Rheumatol Rep.
8:215–223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Becker MA and Jolly M: Hyperuricemia and
associated diseases. Rheum Dis Clin North Am. 32:275–293. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mikuls TR and Saag KG: New insights into
gout epidemiology. Curr Opin Rheumatol. 18:199–203. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Masseoud D, Rott K, Liu-Bryan R and
Agudelo C: Overview of hyperuricaemia and gout. Curr Pharm Des.
11:4117–4124. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi HK, Ford ES, Li C and Curhan G:
Prevalence of the metabolic syndrome in patients with gout: the
Third National Health and Nutrition Examination Survey. Arthritis
Rheum. 57:109–115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi HK, De Vera MA and Krishnan E: Gout
and the risk of type 2 diabetes among men with a high
cardiovascular risk profile. Rheumatology (Oxford). 47:1567–1570.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taniguchi A and Kamatani N: Control of
renal uric acid excretion and gout. Curr Opin Rheumatol.
20:192–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shima Y, Teruya K and Ohta H: Association
between intronic SNP in urate-anion exchanger gene, SLC22A12, and
serum uric acid levels in Japanese. Life Sci. 79:2234–2237. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vázquez-Mellado J, Jiménez-Vaca AL,
Cuevas-Covarrubias S, Alvarado-Romano V, Pozo-Molina G and
Burgos-Vargas R: Molecular analysis of the SLC22A12 (URAT1) gene in
patients with primary gout. Rheumatology (Oxford). 46:215–219.
2007.PubMed/NCBI
|
|
13
|
Miao ZM, Zhao SH, Yan SL, Li CG, Wang YG,
Meng DM, Zhou L and Mi QS: NALP3 inflammasome functional
polymorphisms and gout susceptibility. Cell Cycle. 8:27–30. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miao ZM, Zhao SH, Wang YG, Li CG, Wang ZC,
Chen Y, Chen XY and Yan SL: Epidemiological survey of hyperuricemia
and gout in coastal areas of Shandong Province. Chin J Endocrinol
Metab. 22:421–425. 2006.(In Chinese).
|
|
15
|
Jin C and Flavell RA: Molecular mechanism
of NLRP3 inflammasome activation. J Clin Immunol. 30:628–631. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Agostini L, Martinon F, Burns K, McDermott
MF, Hawkins PN and Tschopp J: NALP3 forms an IL-1beta-processing
inflammasome with increased activity in Muckle-Wells
autoinflammatory disorder. Immunity. 20:319–325. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martinon F and Tschopp J: Inflammatory
caspases: linking an intracellular innate immune system to
autoinflammatory diseases. Cell. 117:561–574. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miao Z, Li C, Chen Y, Zhao S, Wang Y, Wang
Z, Chen X, Xu F, Wang F, Sun R, Hu J, Song W, Yan S and Wang CY:
Dietary and lifestyle changes associated with high prevalence of
hyperuricemia and gout in the Shandong coastal cities of Eastern
China. J Rheumatol. 35:1859–1864. 2008.PubMed/NCBI
|
|
19
|
Martinon F: Mechanisms of uric acid
crystal-mediated autoinflammation. Immunol Rev. 233:218–232. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoffman HM, Mueller JL, Broide DH,
Wanderer AA and Kolodner RD: Mutation of a new gene encoding a
putative pyrin-like protein causes familial cold autoinflammatory
syndrome and Muckle-Wells syndrome. Nat Genet. 29:301–305. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Church LD, Cook GP and McDermott MF:
Primer: inflammasomes and interleukin 1beta in inflammatory
disorders. Nat Clin Pract Rheumatol. 4:34–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pétrilli V and Martinon F: The
inflammasome, autoinflammatory diseases, and gout. Joint Bone
Spine. 74:571–576. 2007.PubMed/NCBI
|
|
23
|
Masters SL, Lobito AA, Chae J and Kastner
DL: Recent advances in the molecular pathogenesis of hereditary
recurrent fevers. Curr Opin Allergy Clin Immunol. 6:428–433. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stojanov S and Kastner DL: Familial
autoinflammatory diseases: genetics, pathogenesis and treatment.
Curr Opin Rheumatol. 17:586–599. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kastner DL: Hereditary periodic fever
syndromes. Hematology Am Soc Hematol Educ Program. 74–81. 2005.
View Article : Google Scholar
|
|
26
|
Wallace SL, Robinson H, Masi AT, Decker
JL, McCarty DJ and Yü TF: Preliminary criteria for the
classification of the acute arthritis of primary gout. Arthritis
Rheum. 20:895–900. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barrett JC, Fry B, Maller J and Daly MJ:
Haploview: analysis and visualization of LD and haplotype maps.
Bioinformatics. 21:263–265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakamura Y, Kambe N, Saito M, Nishikomori
R, Kim YG, Murakami M, Núñez G and Matsue H: Mast cells mediate
neutrophil recruitment and vascular leakage through the NLRP3
inflammasome in histamine-independent urticaria. J Exp Med.
206:1037–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mason DR, Beck PL and Muruve DA:
Nucleotide-binding oligomerization domain-like receptors and
inflammasomes in the pathogenesis of non-microbial inflammation and
diseases. J Innate Immun. 4:16–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang CS, Shin DM and Jo EK: The Role of
NLR-related Protein 3 Inflammasome in Host Defense and Inflammatory
Diseases. Int Neurourol J. 16:2–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Villani AC, Lemire M, Fortin G, Louis E,
Silverberg MS, et al: Common variants in the NLRP3 region
contribute to Crohn’s disease susceptibility. Nat Genet. 41:71–76.
2009.
|
|
32
|
Vandanmagsar B, Youm YH, Ravussin A,
Galgani JE, Stadler K, et al: The NLRP3 inflammasome instigates
obesity-induced inflammation and insulin resistance. Nat Med.
17:179–188. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Serlin DM, Kuang PP, Subramanian M,
O’Regan A, Li X, Berman JS and Goldstein RH: Interleukin-1beta
induces osteopontin expression in pulmonary fibroblasts. J Cell
Biochem. 97:519–529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qu Y, Ramachandra L, Mohr S, Franchi L,
Harding CV, Nunez G and Dubyak GR: P2X7 receptor-stimulated
secretion of MHC class II-containing exosomes requires the
ASC/NLRP3 inflammasome but is independent of caspase-1. J Immunol.
182:5052–5062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eisenbarth SC, Colegio OR, O’Connor W,
Sutterwala FS and Flavell RA: Crucial role for the Nalp3
inflammasome in the immunostimulatory properties of aluminium
adjuvants. Nature. 453:1122–1126. 2008. View Article : Google Scholar
|
|
36
|
Wynn TA: Fibrotic disease and the
T(H)1/T(H)2 paradigm. Nat Rev Immunol. 4:583–594. 2004. View Article : Google Scholar : PubMed/NCBI
|