Introduction
There is increasing evidence that supports the
theory of an inverse association between plasma estrogen levels and
the incidence of cardiovascular disease. The consensus is that
estrogen attenuates during the early and advanced stages of
atherosclerotic lesion development (1).
Vascular endothelial and smooth muscle cells are
important cell types implicated in vascular dysfunction and
atherosclerotic plaque development. Estrogens promote endothelial
integrity by stimulating the production of nitric oxide and
prostacyclin, promoting the proliferation, senescence and migration
of endothelial cells, reducing the adhesion of monocytes and
neutrophils to the endothelial monolayer and repressing the
production of reactive oxygen species. Estrogens have also been
shown to inhibit the proliferation and migration of vascular smooth
muscle cells, and reduce their inflammatory activation and
oxidative stress by increasing the expression of antioxidative
enzymes and reducing the expression and activity of NADPH oxidase
(1).
Estrogen levels are dependent on the activities of
aromatase, 17β-hydroxysteroid dehydrogenase (17β-HSD) type 1,
steroid sulfatase and estrogen sulfotransferase (SULT1E1) (2). Human SULT1E1 is highly efficient at
catalyzing the sulfoconjugation of estrone and estradiol at the
3-hydroxyl terminal. As a sulfotransferase (a group of phase II
drug-metabolizing enzymes), SULT1E1 was observed to affect
tumorigenesis in estrogen-dependent cancers (3), fetal development (4) and adipocyte differentiation (5). Our previous work has demonstrated
that SULT1E1 is highly expressed in human umbilical vein
endothelial cells (HUVECs) and is important in regulating the
inflammatory response and lipid metabolism in endothelial cells
(unpublished data).
Several studies have reported aspects of the
regulation of SULT1E1. Gong et al(6) reported that activation of nuclear
receptor subfamily 3, group C, member 1 (NR3C1; formerly known as
glucocorticoid receptor or GR) by dexamethasone induced the
expression and activity of SULT1E1, and the pregnane X receptor
(PXR) was reported to repress the expression of the SULT1E1
gene via interactions with hepatocyte nuclear factor 4 α (HNF4A) in
human primary hepatocytes and hepatocellular carcinoma cells
(7).
Peroxisome proliferator-activated receptor-α
(PPAR-α), a nuclear receptor that regulates lipid and glucose
metabolism, may affect the development and progression of vascular
lesions (8). A PPAR-α activator
has been observed to reduce the formation of atherosclerotic
lesions in humans and in animal models (9–11).
PPAR-α is expressed in endothelial and vascular smooth muscle
cells, where its effects are anti-inflammatory and
anti-atherogenic, and it also has an inhibitory effect on the
proliferation and migration of vascular cells (12). Fang et al(13) reported that ciprofibrate (a
PPAR-α agonist) increased human SULT2A1
(sulfotransferase family, cytosolic, 2A,
dehydroepiandrosterone-preferring, member 1) mRNA, immunoreactive
protein and enzymatic activity levels by ~2-fold in primary human
hepatocytes. The effect of the PPAR-α agonist on the expression and
activity of SULT1E1 remains unknown.
Insulin-like growth factor-1 (IGF-1) is an endocrine
and autocrine/paracrine growth factor that exists at high levels in
the circulatory system. IGF-1 reduces the atherosclerotic burden
and improves features of atherosclerotic plaque stability in animal
models via multiple potential mechanisms, including by increasing
nitric oxide levels, suppressing plaque cell apoptosis and
downregulating tumor necrosis factor (TNF) (14, 15). The effect of IGF-1 on the
expression and activity of SULT1E1 remains unknown.
As estrogen has a protective role in cardiovascular
disease, and the concentration of estrogen is modulated by SULT1E1,
understanding the factors that regulate the expression and activity
of SULT1E1 is crucial for investigating these diseases. In this
study, we investigated the effects of the PPAR-α agonist WY14643
and IGF-1 on the expression and activity of SULT1E1 in HUVECs and
human umbilical artery smooth muscle cells (HUASMCs).
Materials and methods
Culture of HUVECs and HUASMCs
Written informed consent was obtained from patients
prior to obtaining HUVECs from fresh umbilical cord veins from
normal pregnancies. The study was approved by the ethics committee
of Fudan University in China. HUVECs were isolated by collagenase
digestion, as previously described (16). HUVECs were cultured in an
endothelial cell medium supplemented with 10% fetal bovine serum,
endothelial cell growth supplement and antibiotics. Cells at
passage 1–5 were used in the experiments. The HUVECs were
identified by their typical ‘cobblestone-like’ morphology, positive
immunofluorescence staining for von Willebrand factor and the
uptake of acetylated low-density lipoprotein.
HUASMCs from explants of the human umbilical artery
were cultured in M199 medium supplemented with 10% fetal bovine
serum and antibiotics (17). Cells
were confirmed as smooth muscle cells by their typical
‘hill-and-valley’ morphology and positive immunofluorescent
staining for α-smooth muscle actin (α-SMA). HUASMCs between
passages 3 and 7 were used in all experiments. The cells were
treated with varying doses of the PPAR-α agonist WY14643
(Sigma-Aldrich, St. Louis, MO, USA) dissolved in DMSO, and IGF-1
(PeproTech, London, UK) dissolved in ddH2O.
Immunofluorescence detection of SULT1E1
in HUASMCs
HUASMCs were cultured in collagen-coated glass
bottom dishes. Cells were washed with PBS, fixed with 3.7%
formaldehyde in PBS for 30 min at 4°C and rinsed three times with
PBS at room temperature. Cells were permeabilized in PBS containing
0.2% Triton X-100 and washed with PBS prior to blocking via
incubation with 5% normal goat serum in PBS for 60 min at room
temperature. Cells were subsequently incubated with 2.5% normal
donkey serum in PBS containing SULT1E1 antibody (1:100 dilution;
Proteintech Group, Chicago, IL, USA) or α-SMA antibody (1:100
dilution; Boster Biological Technology., Ltd., Fremont, CA, USA)
overnight at 4°C in an incubator. Cells were washed with PBS
containing 0.05% Tween-20 (3×10 min) and the bound primary
antibodies were visualized with fluorescein isothiocyanate (FITC)
donkey anti-rabbit IgG or Rhodamine TRITC donkey anti-mouse IgG
(Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA).
After washing, the glass bottom dishes were viewed under a
fluorescence microscope. Normal rabbit or mouse IgG served as
negative controls.
Determination of SULT1E1 mRNA levels by
RT-qPCR assay
Total RNA was extracted using an SV Total RNA
Isolation kit (Promega, Madison, WI, USA) according to the
manufacturer’s instructions. Specific mRNA levels were determined
by RT-qPCR assay, as previously described (18). Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as an internal standard.
Specific primer pairs in the experiment are listed in Table I and referenced in PrimerBank
(19).
 | Table IPrimer pairs used to amplify PCR
products. |
Table I
Primer pairs used to amplify PCR
products.
| NCBI gene ID | Gene name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| NM_002046.3 | GAPDH |
TGTTGCCATCAATGACCCCTT |
CTCCACGACGTACTCAGCG |
| NM_005420.2 | SULT1E1 |
GGAAGCCATCAGAGGAGC |
AAAGTGATTTTTCCAGTCTCC |
Determination of SULT1E1 protein levels
by western blot analysis
Following treatment, cells were harvested in
radioimmunoprecipitation (RIPA) lysis buffer. Proteins were
separated by 10% SDS-PAGE in order to detect the SULT1E1 protein
level. β-actin was used as the control. The band densities were
quantified by densitometry using the GIS software (Bio-Tanon,
Shanghai, China).
Determination of SULT1E1 activity by
estrogen sulfation assay
The assay was performed as described by Kushida
et al(20). HUASMCs were
cultured in 24-well plates with 500 μl culture medium, and treated
with PPAR-α agonist WY14643 or IGF-1 for 24 h. Subsequently, the
cells were incubated with 10, 20, 30, 40, 50, 60, 70, 80, 90 or 100
nM cold estrogen combined with 10 nM 3H-labeled
estradiol (E2; 40 Ci/mmol; PerkinElmer, Waltham, MA,
USA) for the sulfation assay. Aliquots (50 μl) of the medium were
sampled at 6 or 24 h, and 50 μl of 0.25 M Tris-HCl (pH 8.7) and 1
ml of water-saturated chloroform were added to the samples.
Following mixing and centrifugation, aliquots (50 μl) of the
aqueous phase (E2S) were collected for liquid
scintillation counting. SULT1E1 activity was represented by the
counts of the aqueous phase.
Construction of human SULT1E1 promoter
luciferase reporter vector
To construct the human SULT1E1
promoter-reporter plasmid, genomic DNA was extracted from the
HUVECs and amplified by PCR to generate a 1246 bp fragment of the
human SULT1E1 5′-flanking sequence. This region spans from
positions −1183 to +62, relative to the upstream region of the
transcription start site. The 5′ promoter region of the
SULT1E1 gene was replicated with the following primers and
subcloned into a luciferase expression vector (pGL3 - basic vector,
Promega) between the KpnI and XhoI restriction sites:
forward, 5′-GGGGTACCCCTTTTGCATAAGGCTAGATA TTG-3′; and reverse,
5′-CCCTCGAGGGTCTCTTCAAATACCAAGGCAG-3′. The sequence authenticity of
the promoter-reporter plasmid was confirmed by DNA sequencing.
Transient transfection
Promoter-reporter plasmids for transient
transfection studies were isolated using a Qiagen plasmid
purification kit (Qiagen, Minneapolis, MN, USA). human embryonic
kidney 293 (HEK-293) cells were cultured in 6-well plates. Cells
were transiently transfected with promoter-reporter plasmids by
Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA), according to
the manufacturer’s instructions. A pRL-CMV plasmid was
co-transfected as a transfection efficiency control. The cells were
treated, after 24 h, by WY14643 or IGF-1 for a further 24 h.
Firefly luciferase and Renilla luciferase activities were
quantified using a Dual-Luciferase® reporter assay
system (Promega). The relative activity fold-changes were
calculated from the results of three independent experiments.
Statistical analysis
Data from three or more separate experiments are
presented as the mean ± SEM. ANOVA and the independent-samples
t-test were performed by SPSS 11.0 software (SPSS Inc., Chicago,
IL, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
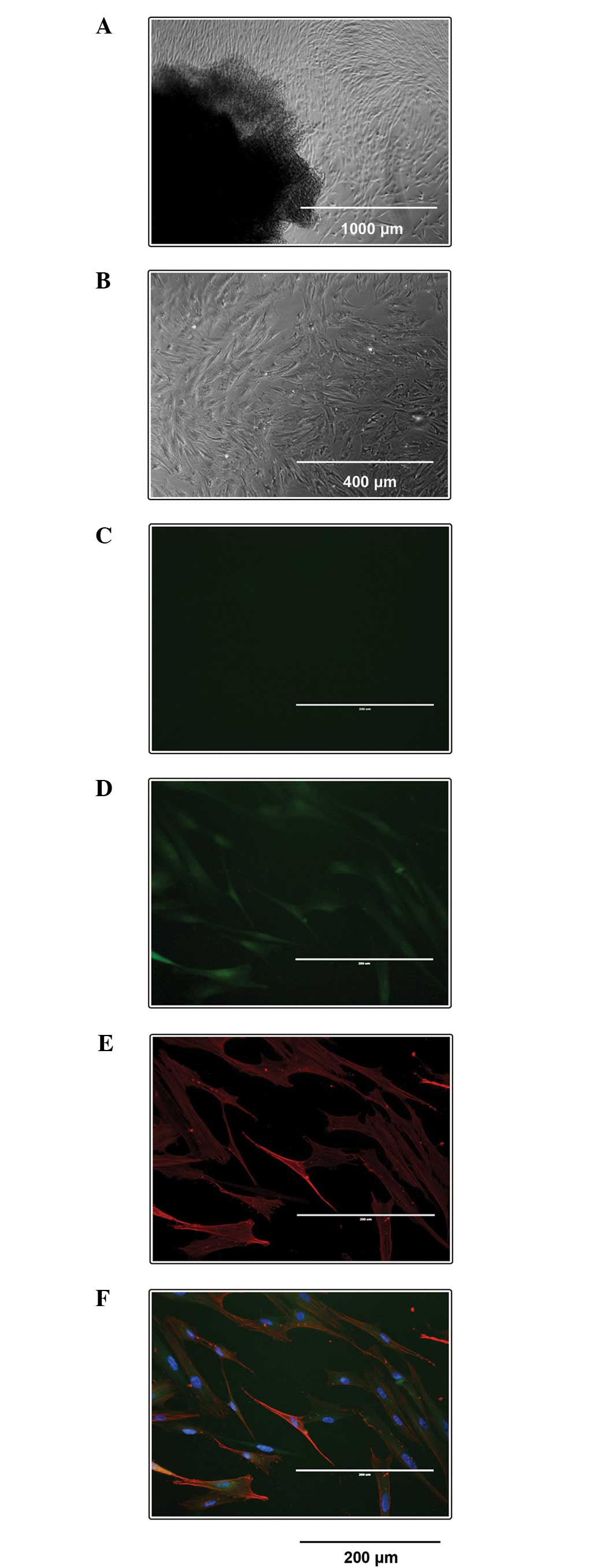
SULT1E1 was highly expressed in
HUASMCs
SULT1E1 expression in HUASMCs was visualized via
immunofluorescence. All HUASMCs demonstrated a typical spindle
morphology with a ‘hill-and-valley’ growth pattern (Fig. 1A and B) and had positive reactions
to the antibody against α-SMA antibody (Fig. 1E). The negative control with normal
rabbit IgG exhibited no staining (Fig.
1C). SULT1E1 was highly expressed in HUASMCs (Fig. 1D).
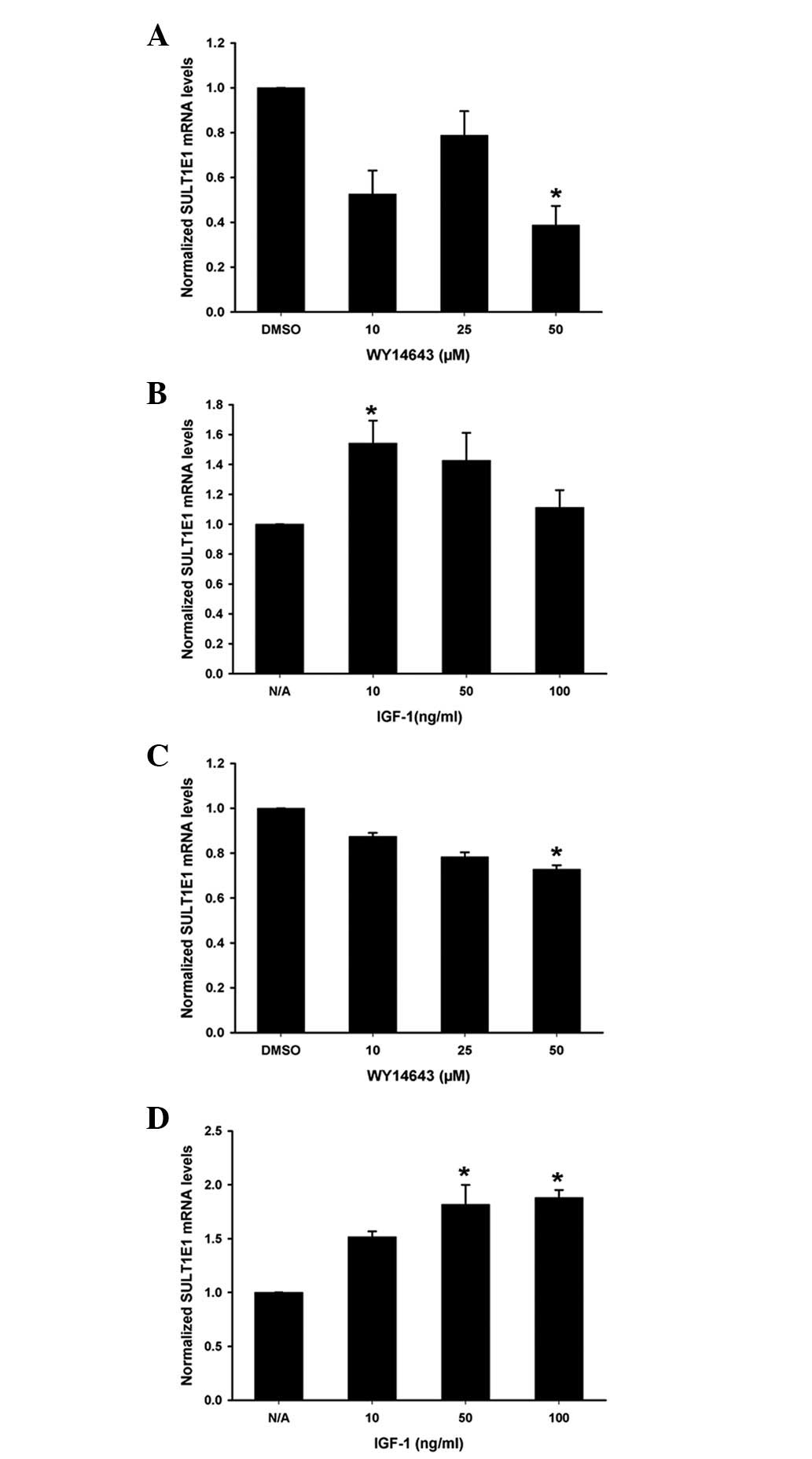
Effects of PPAR-α agonist WY14643 and
IGF-1 on SULT1E1 mRNA levels in HUVECs and HUASMCs
Different concentrations of WY14643 and IGF-1 were
added to the HUVECs (Fig. 2A and
B) or HUASMCs (Fig. 2C and D)
for 24 h. SULT1E1 mRNA levels were measured by RT-qPCR
analysis. WY14643 decreased the SULT1E1 mRNA levels in the
HUVECs and the HUASMCs (Fig. 2A and
C); while IGF-1 significantly increased the SULT1E1 mRNA
levels (Fig. 2B and D).
Effects of PPAR-α agonist WY14643 and
IGF-1 on SULT1E1 protein levels in HUVECs and HUASMCs
In order to confirm the regulatory role of PPAR-α
agonist WY14643 and IGF-1 in SULT1E1 expression, SULT1E1 protein
levels were analyzed using western blotting. Varying concentrations
of PPAR-α agonist WY14643 and IGF-1 were added to the HUVECs
(Fig. 3A-D) or HUASMCs (Fig. 3E-H) for 24 h. The expression of
SULT1E1 was downregulated by WY14643 (Fig. 3A and C), but upregulated by IGF-1,
in the HUVECs (Fig. 3B and D). The
expression pattern of SULT1E1 protein levels in HUASMCs was similar
to that in HUVECs following treatment with the PPAR-α agonist
WY14643 (Fig. 3E and G) and IGF-1
(Fig. 3F and H).
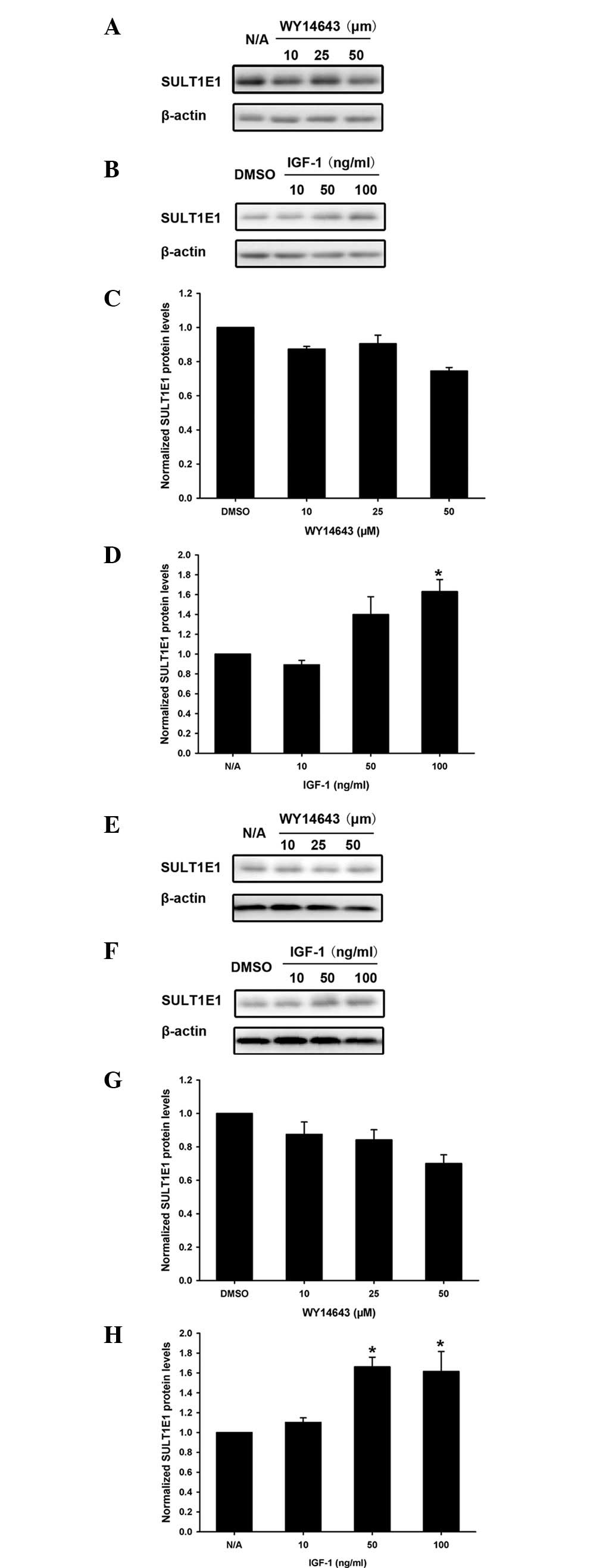 | Figure 3Effects of PPAR-α agonist WY14643 and
IGF-1 on the SULT1E1 protein levels in HUVECs and HUASMCs. (A-D)
HUVECs and (E-H) HUASMCs were treated with different doses of (A,
C, E and G) PPAR-α agonist WY14643 or (B, D, F and H) IGF-1 for 24
h, respectively. The protein levels of SULT1E1 were quantified
using western blot analysis. *P<0.05 compared with
the DMSO or N/A group. PPAR-α, peroxisome proliferator-activated
receptor-α; IGF-1, insulin-like growth factor-1; SULTIE1, estrogen
sulfotransferase; HUVECs, human umbilical vein endothelial cells;
HUASMCs, human umbilical artery smooth muscle cells. |
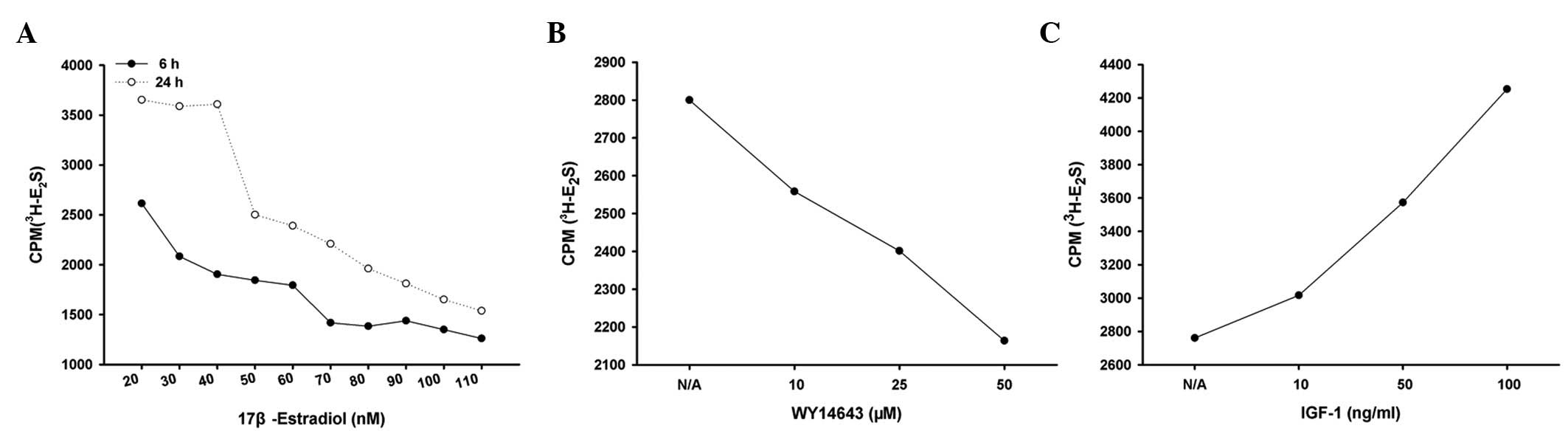
Effects of PPAR-α agonist WY14643 and
IGF-1 on the activity of SULT1E1 in HUASMCs
SULT1E1 activity was measured following treatment
with WY14643 or IGF-1 in HUASMCs. As shown in Fig. 4A, 40 nM estradiol (30 nM estradiol
and 10 nM 3H-estradiol) was capable of complete
sulfation to E2S in untreated HUASMCs following
incubation with 3H-estradiol for 24 h. However, 40 nM
estradiol was a sufficient quantity of substrate for estradiol
sulfation within a 6 h incubation period. Therefore, 6 h was
selected as the incubation period for the subsequent experiments.
HUASMCs were treated with varying concentrations of WY14643 or
IGF-1 for 24 h and were then incubated with 40 nM estradiol (30 nM
estradiol and 10 nM 3H-estradiol) for a further 6 h. The
sulfated estradiol decreased in cells treated with WY14643, but
increased in the presence of IGF-1 (Fig. 4B and C), which suggests that
WY14643 inhibited, whereas IGF-1 enhanced, the activity of SULT1E1
in the HUASMCs.
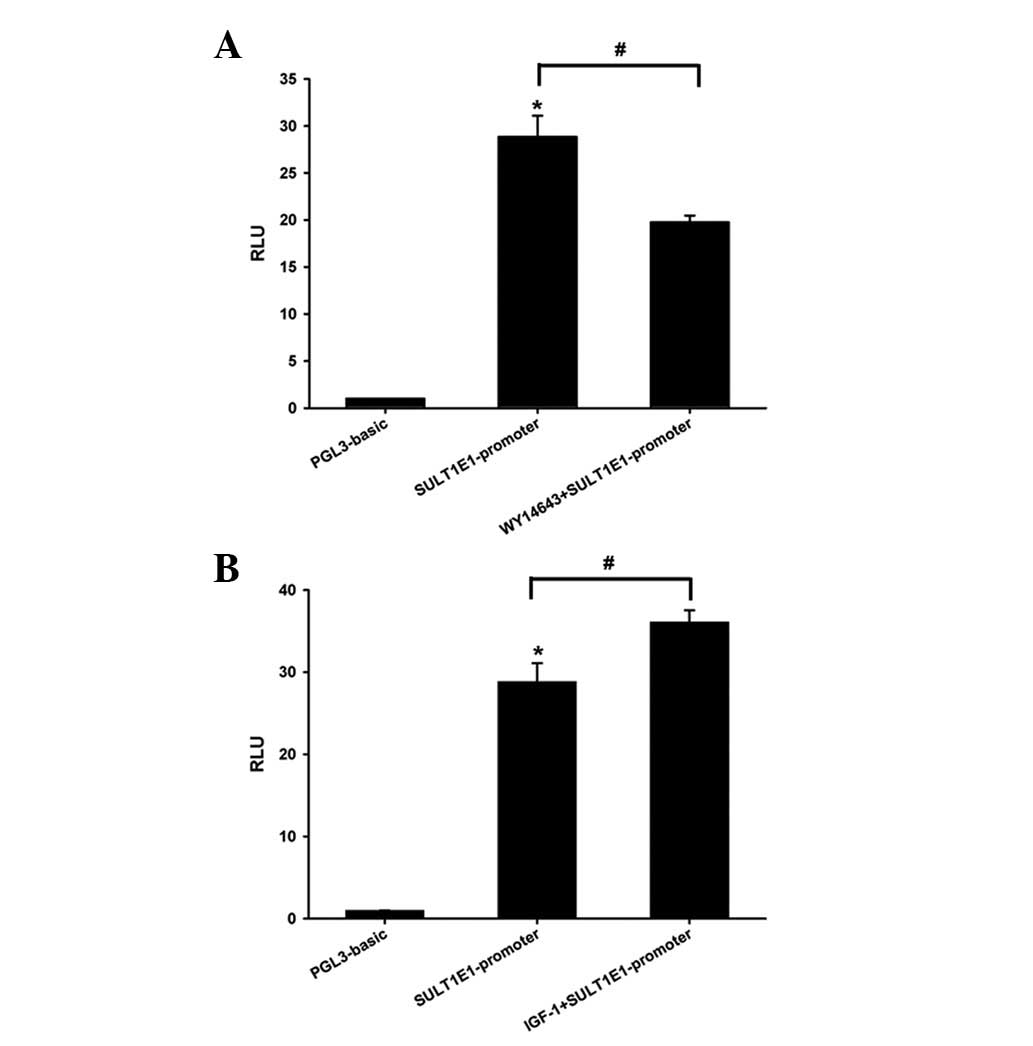
Effect of the PPAR-α agonist WY14643 and
IGF-1 on the activity of the SULT1E1 promoter in HEK-293 cells
In order to clarify the regulatory mechanism of
WY14643 and IGF-1 on SULT1E1 activity, the luciferase assay was
performed using a Dual-Luciferase reporter assay system in HEK-293
cells. The activity of the SULT1E1 promoter increased 30-fold
compared with the pGL3-basic vector. PPAR-α agonist WY14643
decreased, while IGF-1 increased, the luciferase activity of the
SULT1E1 promoter (Fig. 5). The
data suggested that WY14643 and IGF-1 regulated SULT1E1 expression
at the transcriptional level.
Discussion
Estrogens exert potent atheroprotective effects by
attenuating vascular dysfunctions. Estrogen metabolism is involved
in the regulation of estrogen activity and affects vascular
function. SULT1E1 is the predominant enzyme responsible for the
sulfation of β-estradiol and estrone (21). According to our previous studies
and the present study, SULT1E1 is abundantly expressed in HUVECs
and HUASMCs. This suggests that vascular function is associated
with the expression and activity of SULT1E1. This study
demonstrated that the PPAR-α agonist WY14643 downregulated, but
IGF-1 upregulated, the expression and activity of SULT1E1 in
vascular cells. Since the SULT1E1 mRNA levels and luciferase
activity of the SULT1E1 promoter were decreased by WY14643 and
increased by IGF-1 treatment, we hypothesize that the PPAR-α
agonist WY14643 and IGF-1 regulated SULT1E1 expression at the
transcriptional level.
Increasing amounts of evidence indicate that PPAR-α
agonists improve vascular function independent of their
hypolipidemic effects (12). The
underlying mechanisms include inhibiting inflammatory cell
recruitment and activation, attenuating inflammatory responses
(22), increasing cholesterol
efflux and plaque stability, decreasing foam cell formation
(23), vasoconstriction and
thrombosis, and controlling vascular smooth muscle cell
proliferation (24). In this
study, we observed that the PPAR-α agonist WY14643 inhibited the
expression and activity of SULT1E1 in vascular cells. Therefore,
the PPAR-α agonist may increase the concentration and activity of
estrogen by reducing the expression of SULT1E1. Less 17β-estradiol
was sulfated in HUASMCs treated with WY14643 compared with
untreated cells.
Activation of PPAR-α is initiated by the binding of
an agonist. Following ligand activation, PPAR-α forms heterodimers
with retinoid X receptor (RXR). The PPAR-RXR complex subsequently
recognizes and binds to PPAR-response elements (PPREs) on target
gene promoters, thereby activating their expression (25). Furthermore, the activation of the
PPAR-RXR complex participates in the negative regulation of certain
genes, either by processes independent of DNA-binding via
interference with other signaling pathways, or by a DNA-dependent
process involving the recruitment of co-repressors (26). Based on the data of the RT-qPCR
assay and SULT1E1 promoter reporter activity assay, WY14643
regulated the expression and activity of SULT1E1 in vascular
cells at the transcriptional level. However, the precise molecular
mechanism of this repression requires further investigation.
IGF-1 exerts an anti-atherogenic effect. In
endothelial cells, IGF-1 enhances the production of nitric oxide by
endothelial nitric oxide synthase (eNOS) (27). In smooth muscle cells, IGF-1
prevents oxidative stress-induced apoptotic cell death and
stimulates smooth muscle cell proliferation, migration and
extracellular matrix synthesis, indicating a role for IGF-1 in the
maintenance of plaque stability (28). According to our data, SULT1E1
expression and activity were enhanced by IGF-1. Once IGF-1 reduces
the concentration of estrogen by increasing SULT1E1 activity, the
anti-atherogenic role of estrogen may be attenuated. The actions of
IGF-1 are mediated by the specific membrane receptor IGF-1R, a
tyrosine kinase that undergoes autophosphorylation and catalyzes
the phosphorylation of insulin receptor substrate proteins. Upon
phosphorylation, insulin receptor substrate proteins interact with
signaling molecules, including Akt, Ras/Raf and Rac (29). The precise molecular mechanism that
underlies the increase in SULT1E1 expression by IGF-1 at the
transcriptional level requires further investigation.
In conclusion, this study investigated the effects
of two important mediators of the process of atherosclerosis, a
PPAR-α agonist and IGF-1, on the expression and activity of SULT1E1
in vascular cells. Thus, the PPAR-α agonist and IGF-1 may regulate
the levels of active estrogens in endothelial cells and smooth
muscle cells, thereby affecting the physiology and pathophysiology
of the vascular walls.
Acknowledgements
This work was supported by The National Natural
Science Foundation of China (NSFC 81030014, NSFC 81270497 and NSFC
81101762).
References
|
1
|
Nofer JR: Estrogens and atherosclerosis:
insights from animal models and cell systems. J Mol Endocrinol.
48:R13–R29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ishibashi H, Suzuki T, Suzuki S, et al:
Estrogen inhibits cell proliferation through in situ production in
human thymoma. Clin Cancer Res. 11:6495–6504. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu Y, Liu X, Guo F, et al: Effect of
estrogen sulfation by SULT1E1 and PAPSS on the development of
estrogen-dependent cancers. Cancer Sci. 103:1000–1009. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tong MH, Jiang H, Liu P, Lawson JA, Brass
LF and Song WC: Spontaneous fetal loss caused by placental
thrombosis in estrogen sulfotransferase-deficient mice. Nat Med.
11:153–159. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wada T, Ihunnah CA, Gao J, et al: Estrogen
sulfotransferase inhibits adipocyte differentiation. Mol
Endocrinol. 25:1612–1623. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gong H, Jarzynka MJ, Cole TJ, et al:
Glucocorticoids antagonize estrogens by glucocorticoid
receptor-mediated activation of estrogen sulfotransferase. Cancer
Res. 68:7386–7393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kodama S, Hosseinpour F, Goldstein JA and
Negishi M: Liganded pregnane X receptor represses the human
sulfotransferase SULT1E1 promoter through disrupting its chromatin
structure. Nucleic Acids Res. 39:8392–8403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fruchart JC: Peroxisome
proliferator-activated receptor-alpha (PPARalpha): at the
crossroads of obesity, diabetes and cardiovascular disease.
Atherosclerosis. 205:1–8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hennuyer N, Tailleux A, Torpier G, et al:
PPARalpha, but not PPARgamma, activators decrease macrophage-laden
atherosclerotic lesions in a nondiabetic mouse model of mixed
dyslipidemia. Arterioscler Thromb Vasc Biol. 25:1897–1902. 2005.
View Article : Google Scholar
|
|
10
|
Srivastava RA: Evaluation of
anti-atherosclerotic activities of PPAR-alpha, PPAR-gamma, and LXR
agonists in hyperlipidemic atherosclerosis-susceptible F(1)B
hamsters. Atherosclerosis. 214:86–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zahradka P, Yurkova N, Litchie B, Moon MC,
Del Rizzo DF and Taylor CG: Activation of peroxisome
proliferator-activated receptors alpha and gamma1 inhibits human
smooth muscle cell proliferation. Mol Cell Biochem. 246:105–110.
2003. View Article : Google Scholar
|
|
12
|
Hamblin M, Chang L, Fan Y, Zhang J and
Chen YE: PPARs and the cardiovascular system. Antioxid Redox
Signal. 11:1415–1452. 2009. View Article : Google Scholar
|
|
13
|
Fang HL, Strom SC, Cai H, Falany CN,
Kocarek TA and Runge-Morris M: Regulation of human hepatic
hydroxysteroid sulfotransferase gene expression by the peroxisome
proliferator-activated receptor alpha transcription factor. Mol
Pharmacol. 67:1257–1267. 2005. View Article : Google Scholar
|
|
14
|
Higashi Y, Sukhanov S, Anwar A, Shai SY
and Delafontaine P: IGF-1, oxidative stress and atheroprotection.
Trends Endocrinol Metab. 21:245–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Higashi Y, Sukhanov S, Anwar A, Shai SY
and Delafontaine P: Aging, atherosclerosis, and IGF-1. J Gerontol A
Biol Sci Med Sci. 67:626–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han Y, Qi Y, Kang J, Li N, Tian X and Yan
C: Nerve growth factor promotes formation of lumen-like structures
in vitro through inducing apoptosis in human umbilical vein
endothelial cells. Biochem Biophys Res Commun. 366:685–691. 2008.
View Article : Google Scholar
|
|
17
|
Vadiveloo PK, Filonzi EL, Stanton HR and
Hamilton JA: G1 phase arrest of human smooth muscle cells by
heparin, IL-4 and cAMP is linked to repression of cyclin D1 and
cdk2. Atherosclerosis. 133:61–69. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yeong P, Ning Y, Xu Y, Li X and Yin L:
Tryptase promotes human monocyte-derived macrophage foam cell
formation by suppressing LXRalpha activation. Biochim Biophys Acta.
1801:567–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Spandidos A, Wang H and Seed B:
PrimerBank: a PCR primer database for quantitative gene expression
analysis, 2012 update. Nucleic Acids Res. 40:D1144–D1149. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kushida A, Hattori K, Yamaguchi N,
Kobayashi T, Date A and Tamura H: Sulfation of estradiol in human
epidermal keratinocyte. Biol Pharm Bull. 34:1147–1151. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu Y, Yang X, Wang Z, et al: Estrogen
sulfotransferase (SULT1E1) regulates inflammatory response and
lipid metabolism of human endothelial cells via PPARgamma. Mol Cell
Endocrinol. 369:140–149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lalloyer F, Wouters K, Baron M, et al:
Peroxisome proliferatoractivated receptor-alpha gene level
differently affects lipid metabolism and inflammation in
apolipoprotein E2 knock-in mice. Arterioscler Thromb Vasc Biol.
31:1573–1579. 2011. View Article : Google Scholar
|
|
23
|
Chinetti G, Lestavel S, Bocher V, et al:
PPAR-alpha and PPAR-gamma activators induce cholesterol removal
from human macrophage foam cells through stimulation of the ABCA1
pathway. Nat Med. 7:53–58. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gizard F, Nomiyama T, Zhao Y, et al: The
PPARalpha/p16INK4a pathway inhibits vascular smooth muscle cell
proliferation by repressing cell cycle-dependent telomerase
activation. Circ Res. 103:1155–1163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Plutzky J: The PPAR-RXR transcriptional
complex in the vasculature: energy in the balance. Circ Res.
108:1002–1016. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zoete V, Grosdidier A and Michielin O:
Peroxisome proliferator-activated receptor structures: ligand
specificity, molecular switch and interactions with regulators.
Biochim Biophys Acta. 1771:915–925. 2007. View Article : Google Scholar
|
|
27
|
Sukhanov S, Higashi Y, Shai SY, et al:
Differential requirement for nitric oxide in IGF-1-induced
anti-apoptotic, anti-oxidant and anti-atherosclerotic effects. FEBS
Lett. 585:3065–3072. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Razuvaev A, Folkersen L, et al:
The expression of IGFs and IGF binding proteins in human carotid
atherosclerosis, and the possible role of IGF binding protein-1 in
the regulation of smooth muscle cell proliferation.
Atherosclerosis. 220:102–109. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Delafontaine P, Song YH and Li Y:
Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1
binding proteins in blood vessels. Arterioscler Thromb Vasc Biol.
24:435–444. 2004. View Article : Google Scholar : PubMed/NCBI
|