Introduction
Acute pancreatitis, particularly the necrotizing
type, is associated with high morbidity and mortality. Necrotizing
pancreatitis is characterized by parenchymal non-enhancement on
contrast-enhanced computed tomography images (1). Although necrosis is irreversible,
certain patients exhibiting this pancreatic parenchymal
non-enhancement recover with normal pancreatic conditions (2). Vasospasm has been implicated in the
development of pancreatic ischemia and necrosis (3); however, the precise underlying
mechanism is unclear.
The local pancreatic renin-angiotensin system (RAS)
may be a significant etiological factor of pancreatitis (4–9).
Angiotensin II receptors have been detected in the endothelium of
blood vessels and the epithelium of the pancreatic ductal system
(8). Previous studies have
revealed that experimental pancreatitis upregulates the RAS
components in the pancreas (10,11).
However, elevated local angiotensin II levels in pancreatitis
tissues have not been reported. Trypsin may directly induce the
generation of the pressor angiotensin II from angiotensinogen at a
weakly acidic pH, independent of the action of
angiotensin-converting enzyme (12,13).
In the present study, pancreatic angiotensin II
concentration was measured in experimental animals with
pancreatitis to evaluate the involvement of the local angiotensin
II generating system in acute pancreatitis.
Materials and methods
Animal and experimental acute
pancreatitis model
The present study was conducted in compliance with
the Division for Animal Research Resources, Institute of Kanazawa
University. The experiments and procedures were approved by the
Animal Care and Use Committee of the Kanazawa University (Ishikawa,
Japan).
Male Wistar rats (weight, 250–350 g) were maintained
on a 12-h light/dark cycle at an ambient temperature of 23°C. The
rats were fasted with free access to water for 12 h prior to
surgery. Rats were anesthetized with diethyl ether inhalation and
laparotomy was performed using a midline abdominal incision. Acute
pancreatitis was induced by retrograde injection of 6% sodium
taurocholate (Wako Pure Chemical Industries, Osaka, Japan) into the
biliopancreatic duct at a dose of 0.4 ml/kg body weight to induce
acute pancreatitis. Time 0 indicates the time of injection of
sodium taurocholate. The rats were divided into groups and
sacrificed at 3, 6, 12, 24 or 36 h following the induction of acute
pancreatitis. Control rats were sacrificed without injection into
the biliopancreatic duct.
Sample collection and preparation
Blood samples were collected by cardiac puncture for
the measurement of plasma amylase levels. Pancreatic tissue samples
were fixed in 10% neutral-buffered formalin and embedded in
paraffin for histological examination. Parallel samples of
pancreatic tissue were prepared for the measurement of angiotensin
II concentration by immediately freezing in liquid nitrogen,
followed by storage at −80°C until the time of assay.
Measurements of angiotensin II in
tissues
Frozen tissue samples were homogenized at 4°C in
saline containing 0.1% NHCl and 5% urinastatin. The homogenate was
centrifuged at 10,000 × g for 30 min at 4°C and the supernatant was
collected for radioimmunoassay of angiotensin II using the florisil
method, as described previously (14).
Statistical analysis
Data were analyzed using the Welch’s t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
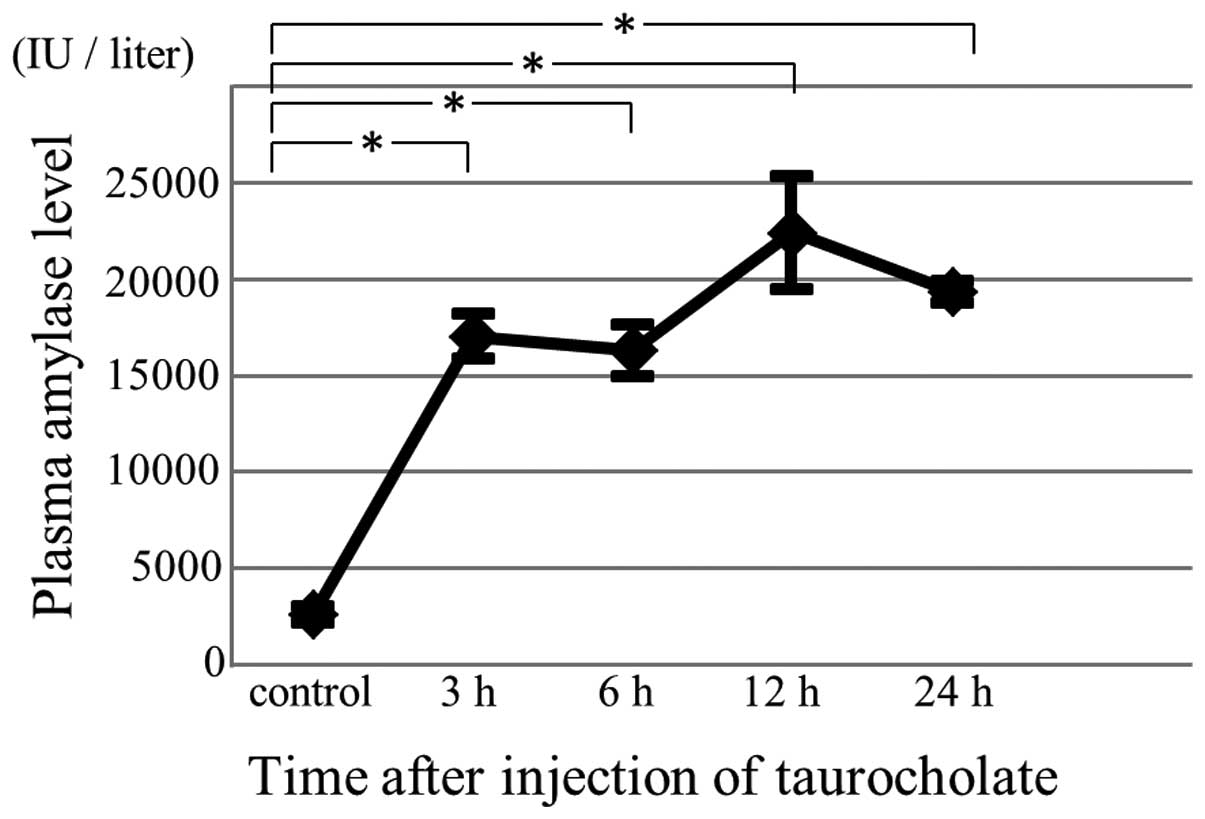
Plasma amylase levels
Plasma amylase levels were measured to confirm the
successful induction of acute pancreatitis. Following this
induction, plasma amylase levels were elevated (Fig. 1).
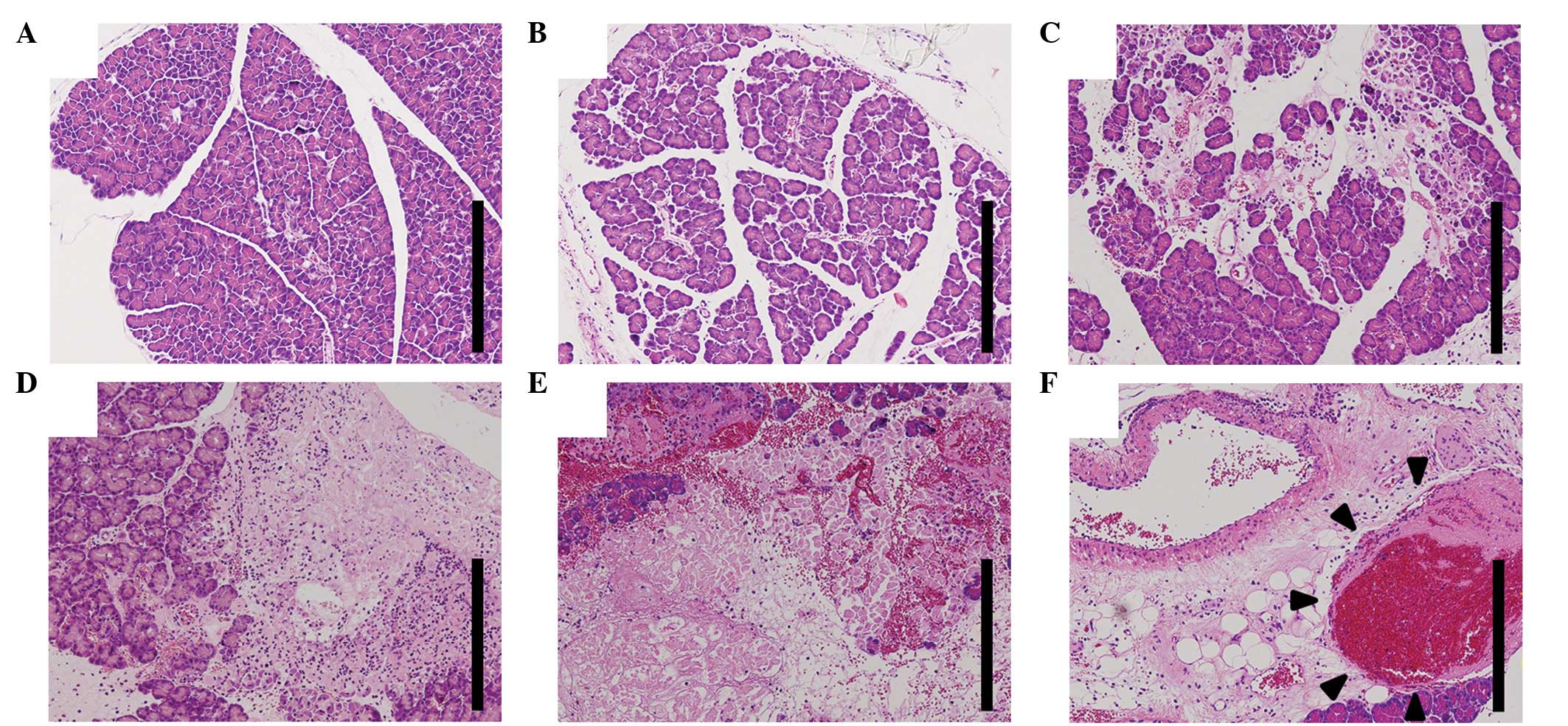
Histological examination
In the early stages of pancreatitis, histological
examination indicated subcapsular and interlobular edema, slight
inflammatory neutrophilic infiltration and a small number of
intralobular necrotic lesions. The extent of inflammatory cell
infiltration and areas of necrosis progressed with time. At 24 h
following taurocholate injection, marked inflammatory and necrotic
changes were noted (Fig. 2A-E). In
addition, intravenous thromboses were observed, but there was no
evidence of arterial thrombosis (Fig.
2F).
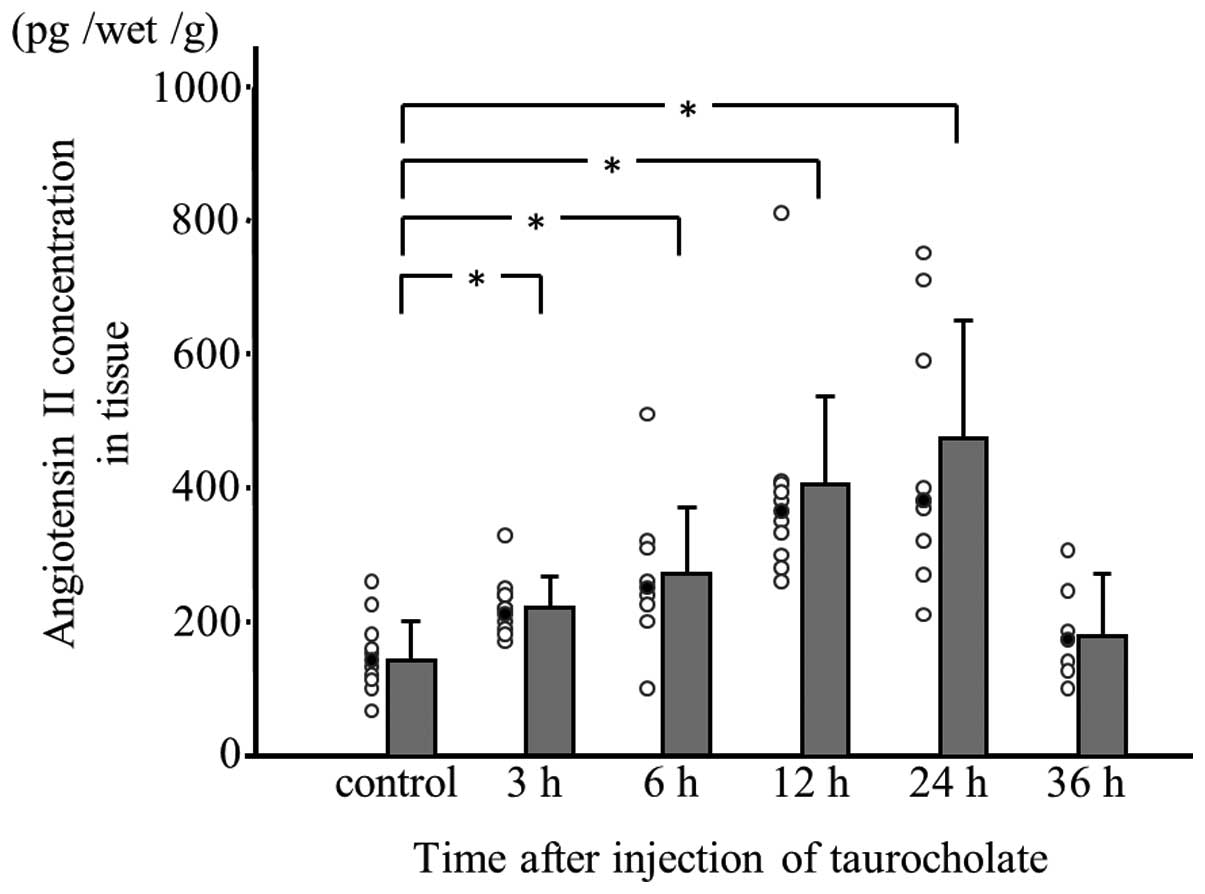
Angiotensin II concentration in
experimental pancreatitis tissue
Angiotensin II concentration was measured in
pancreatic tissue samples obtained at 3, 6, 12, 24 and 36 h
following taurocholate injection. Up to 24 h, the angiotensin II
concentration was significantly higher in rats with
taurocholate-induced pancreatitis compared with controls and the
enzyme levels were found to increase in a time-dependent manner.
However, the angiotensin II concentration in pancreatitis tissue at
36 h following taurocholate injection was lower than that in
pancreatitis tissue at 24 h following injection (Fig. 3).
Discussion
Premature activation of trypsin within pancreatic
acinar cells by various proteases, which in turn are activated by
ectopic activation of trypsinogen, is hypothesized to represent the
main etiological factor in pancreatitis. A number of studies in
acute pancreatitis have examined the mechanisms by which edematous
injury progresses to necrosis. In experimental models, pancreatic
vascular perfusion failure has been implicated in the development
of pancreatitis. Spormann et al(15) demonstrated that temporary ischemia
significantly augmented the severity of pathology and Klar et
al(16) revealed that
vasoconstrictor administration has adverse effects in experimental
acute pancreatitis. Intravascular thromboses have been indicated to
represent a significant factor (17). Although intravenous thromboses were
noted in the present study, arterial thromboses were not
observed.
Deterioration of the balance between nitric oxide
and endothelins has also been hypothesized to account for the
development of necrotizing pancreatitis (18,19);
however, the association between this abnormal balance and trypsin,
the key molecule in the etiology of pancreatitis, is unclear.
It has been hypothesized that an intrinsic tissue
RAS may be present in the pancreas (4). Chan et al(20) have demonstrated activation of the
pancreatic RAS in a rat model of chronic hypoxia. Leung et
al(10) revealed that
experimental pancreatitis induced upregulation of several key RAS
components in the pancreas. Several other studies have reported
that RAS blockers are effective in the treatment of experimental
pancreatitis (21–23).
Hypovascularity is a key characteristic of not only
necrotizing pancreatitis but also pancreatic ductal cancer. The
majority of pancreatic ductal cancers are hypovascular or avascular
on angiography, but the angiography of surgically resected
specimens cannot always accurately confirm poor circulation.
Figarella et al(24)
reported that cationic trypsinogen is spontaneously converted to
trypsin under acidic conditions and Tajima et al(25) demonstrated that tumor-derived
trypsin, activated by acidic conditions, was a key factor in tumor
invasion. Ohta et al(26)
found that angiotensin II acts on the pre-existing pancreatic
arteries around the tumor leading to formation of hypovascular
regions. Since the microenvironment around the tumor cells is more
acidic than that of the majority of normal tissues, this phenomenon
may be associated with tumor-derived trypsin, directly generating
angiotensin II from angiotensinogen (27,28).
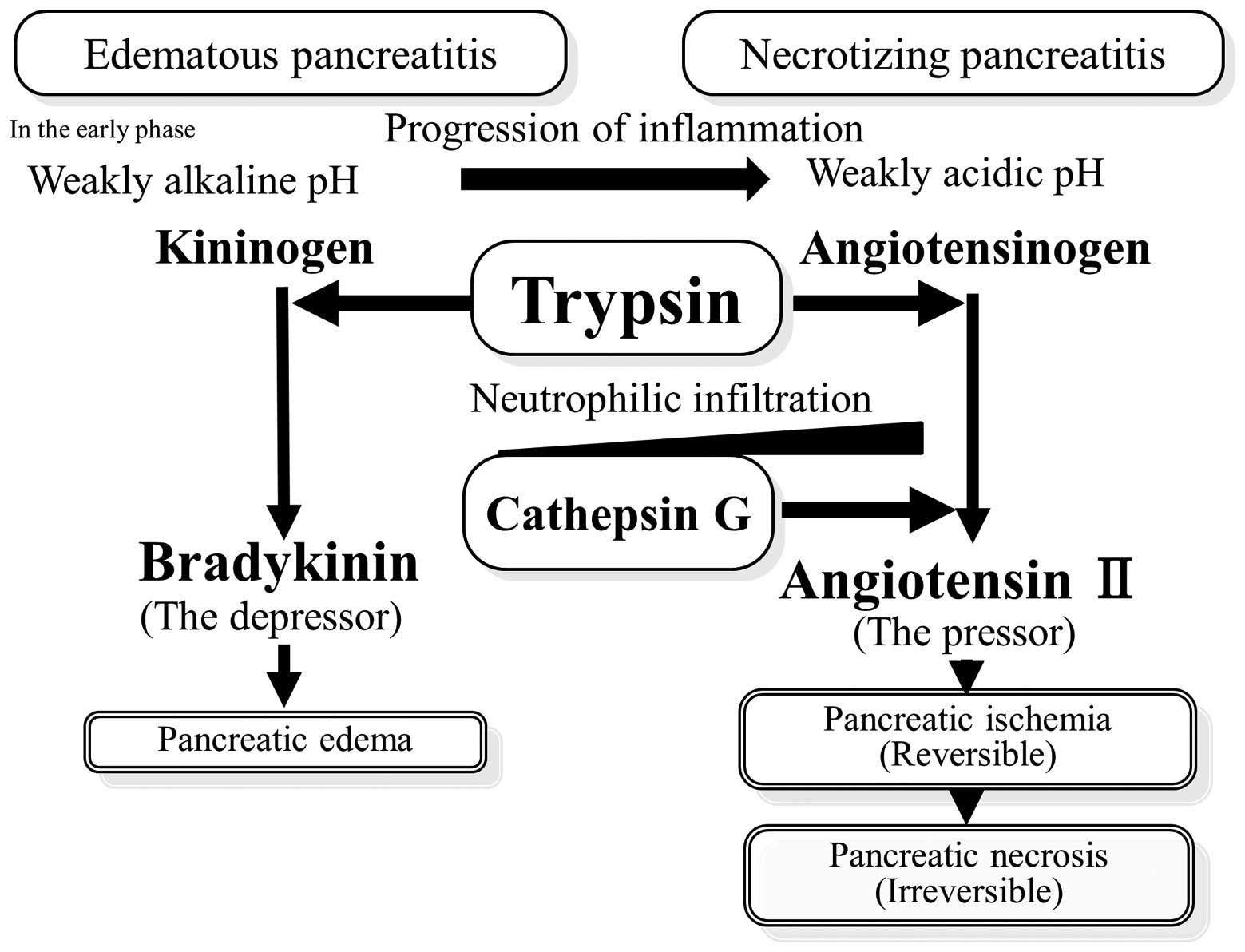
It was reported that the pH of the inflammatory
exudate decreases with time (29–31)
due to local acid production by glycolytic infiltrating neutrophils
and low oxygen tension in inflammatory lesions. In the early phase
of pancreatitis development, the depressor, bradykinin, was
produced at a weakly alkaline pH. Therefore, pancreatic hyperemia
and edema were observed. With time, the inflammation progressed, pH
decreased and angiotensin II was produced under weakly acidic
conditions by trypsin. In addition, pancreatic ischemia and
necrosis were observed (Fig. 4).
In our model of experimental pancreatitis, angiotensin II
concentration decreased at 36 h, as compared with that at 24 h. We
hypothesize that pancreatic tissue destruction may be responsible
for the degradation of angiotensin II.
Cathepsin G, one of the serine proteinases expressed
in the azurophilic granules of neutrophils, has the potential to
induce angiotensin II production (32). In the early phases of acute
pancreatitis, the presence of subcapsular and intralobular edema,
slight neutrophil infiltration and a small number of intralobular
necrotic lesions were noted. The extent of neutrophil infiltration
and areas of necrosis progressed over time. Cathepsin G released by
infiltrating neutrophils may be involved in angiotensin II
generation (33). As the
inflammation progressed with time, angiotensin II was produced by
cathepsin G released by infiltrating neutrophils (Fig. 4). A number of studies have reported
that the neutrophil elastase inhibitor prevents neutrophil
infiltration into the extravascular tissue (34,35).
Although there are no cathepsin G inhibitors available for clinical
use today, the neutrophil elastase inhibitor may suppress the
angiotensin II generated by the cathepsin G released by
infiltrating neutrophils.
In conclusion, the present study indicates that
angiotensin II generation is instrumental in the transition from
experimental edematous pancreatitis to necrotizing pancreatitis. We
hypothesize that locally formed angiotensin II may act on the
pancreatic arteries leading to regional hypovascularity. Therefore,
inhibition of the angiotensin II generating system by
administration of angiotensin receptor blockers, serine protease
inhibitors or elastase inhibitors may be useful for preventing the
evolution of edematous pancreatitis into necrotizing
pancreatitis.
References
|
1
|
Bollen TL, van Santvoort HC, Besselink MG,
et al: The Atlanta Classification of acute pancreatitis revisited.
Br J Surg. 95:6–21. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Traverso LW and Kozarek RA: Pancreatic
necrosectomy: definitions and technique. J Gastrointest Surg.
9:436–439. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takeda K, Mikami Y, Fukuyama S, et al:
Pancreatic ischemia associated with vasospasm in the early phase of
human acute necrotizing pancreatitis. Pancreas. 30:40–49.
2005.PubMed/NCBI
|
|
4
|
Paul M, Poyan Mehr A and Kreutz R:
Physiology of local renin-angiotensin systems. Physiol Rev.
86:747–803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chappell MC, Millsted A, Diz DI, et al:
Evidence for an intrinsic angiotensin system in the carine
pancreas. J Hypertens. 9:751–759. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chappell MC, Jacobsen DW and Tallant EA:
Characterization of angiotensin II receptor subtypes in pancreatic
acinar AR42J cells. Peptides. 16:741–747. 1995. View Article : Google Scholar
|
|
7
|
Ghiani BU and Masini MA: Angiotensin II
binding sites in the rat pancreas and their modulation after
sodium. Comp Biochem Physiol A Physiol. 111:439–444. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leung PS, Chan WP, Wong TP, et al:
Expression and localization of the renin-angiotensin system in the
rat pancreas. J Endocrinol. 160:13–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tahmasebi M, Puddefoot JR, Inwang ER, et
al: The tissue renin-angiotensin system in human pancreas. J
Endocrinol. 161:317–322. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leung PS, Chan WP and Nobiling R:
Regulated expression of pancreatic renin-angiotensin system in
experimental pancreatitis. Mol Cell Endocrinol. 166:121–128. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ip SP, Kwan PC, Williams CH, et al:
Changes of angiotensin-converting enzyme activity in the pancreas
of chronic hypoxia and acute pancreatitis. Int J Biochem Cell Biol.
35:944–954. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arakawa K, Ikeda M, Fukuyama J, et al: A
pressor formation by trypsin from renin-denatured human plasma
protein. J Clin Endocrinol Metab. 42:599–602. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arakawa K and Maruta H: Ability of
kallikrein to generate angiotensin II-like pressor substance and a
proposed ‘kinin-tensin enzyme system’. Nature. 288:705–706.
1980.PubMed/NCBI
|
|
14
|
Morimoto T, Aoyama M, Gotoh E, et al: A
method of radioimmunoassay of plasma angiotensin II using florisil.
Nihon Naibunpi Gakkai Zasshi. 59:15–29. 1983.(In Japanese).
|
|
15
|
Spormann H, Sokolowski A and Letko G:
Effect of temporary ischemia upon development and histological
patterns of acute pancreatitis in the rat. Pathol Res Pract.
184:507–513. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klar E, Rattner DW, Compton C, et al:
Adverse effect of theraeuic vasoconstrictors in experimental acute
pancreatitis. Ann Surg. 214:168–174. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Menger MD, Plusczyk T and Vollmar B:
Microcirculatory derangements in acute pancreatitis. J
Hepatobiliary Pancreat Surg. 8:187–194. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu XH, Kimura T, Ishikawa H, et al:
Effect of endothelin-1 on the decelopment of hemorrhagic
pancreatitis in rats. Scand J Gastroenterol. 30:276–282. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Foitzik T, Faulhaber J, Hotz HG, et al:
Endothelin mediates local and systemic disease sequelae in severe
experimental pancreatitis. Pancreas. 22:248–254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chan WP, Fung ML, Nobiling R, et al:
Activation of local renin-angiotensin system by chronic hypoxia in
rat pancreas. Mol Cell Endocrinol. 160:107–114. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsang SW, Cheng CH and Leung PS: The role
of the pancreatic rennin-angiotensin system in acinar digestive
enzyme secretion and in acute pancreatitis. Regul Pept.
119:213–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chan YC and Leung PS: Angiotensin II type
1 receptor-dependent nuclear factor-kB activation-mediated
proinflammatory actions in a rat model of obstructive acute
pancreatitis. J Pharmacol Exp Ther. 323:10–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oruc N, Ozutemiz O, Nart D, et al:
Inhibition of renin-angiotensin systems in experimental acute
pancreatitis in rats: A new therapeutic target? Exp Toxicol Pathol.
62:353–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Figarella C, Miszczuk-Jamska B and Barrett
AJ: Possible lysosomal activation of pancreatic zymogens.
Activation of both human trypsinogens by cathepsin B and
spontaneous acid Activation of human trypsinogen 1. Biol Chem Hoppe
Seyler. 369:293–298. 1988.PubMed/NCBI
|
|
25
|
Tajima H, Ohta T, Elnemr A, et al:
Enhanced incasiveness of pancreatic adenocarcinoma cells stably
transfected with cationic trypsinogen cDNA. Int J Cancer.
94:699–704. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohta T, Amaya K, Yi S, et al: Angiotensin
converting enzyme-independent, local angiotensin II-generation in
human pancreatic ductal cancer tissue. Int J Oncol. 23:593–598.
2003.PubMed/NCBI
|
|
27
|
Montcourrier P, Silver I, Farnoud R, et
al: Breast cancer cells have a higher capacity to acidify
extracellular milieu by a ductal mechanism. Clin Exp Metastasis.
15:382–392. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ohta T, Numata M, Yagishita H, et al:
Expression of 16 kDa proteolipid of vacuolar-type H(+)-ATPase in
human pancreatic cancer. Br J Cancer. 73:1511–1517. 1996.
|
|
29
|
Donald WE and Walter HS: The pH of
inflammatory exudates. Proc Soc Exp Biol Med. 137:1328–1332. 1971.
View Article : Google Scholar
|
|
30
|
Simmen HP, Battaglia H, Giovanoli P, et
al: Analysis of pH, pO2 and pCO2 in drainage
fluid allows for rapid detection of infectious complications during
the follow-up period after abdominal surgery. Infection.
22:386–389. 1994.PubMed/NCBI
|
|
31
|
Ward TT and Steigbigel RT: Acidosis of
synovial fluid correlates with synovial fluid leukocytosis. Am J
Med. 64:933–936. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wintroub BU, Klickstein LB and Watt KW: A
human neutrophil-dependent pathway for generation of angiotensin
II. Purification of the product and identification as angiotensin
II. J Clin Invest. 68:484–490. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rykl J, Thiemann J, Kuzawski S, et al:
Renal cathepsin G and angiotensin II generation. J Hypertens.
24:1797–1807. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kawabata K, Hagio T, Matsumoto S, et al:
Delayed neutrophil elastase inhibition prevents subsequent
progression of acute lung injury induced by endotoxin inhalation in
hamsters. Am J Respir Crit Care Med. 161:2013–2018. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Young RE, Voisin MB, Wang S, et al: Role
of neutrophil elastase in LTB4-induced neutrophil transmigration in
vivo assessed with a specific inhibitor and neutrophil elastase
deficient mice. Br J Pharmacol. 151:628–637. 2007. View Article : Google Scholar : PubMed/NCBI
|