Introduction
X-linked ichthyosis (XLI; OMIM #308100) is an
inherited metabolic disorder resulting from a steroid sulfatase
(STS) deficiency (1). It affects
~1:2,000–6,000 males, with little racial or geographic variation
(2–4). Of the males affected, >90%
manifest with generalized exfoliation of the skin within a few
weeks of being born. Later, they develop large, polygonal,
dark-brown scales symmetrically distributed over the extremities,
trunk and neck, but typically sparing the face, palms and soles.
The skin lesions persist throughout the life of the patient.
Extracutaneous manifestations are common, particularly with corneal
opacities and cryptorchidism (4).
Other associated features, although extremely rare, include mental
retardation, epilepsy, pyloric hypertrophy, congenital defects of
the abdominal wall and acute lymphoblastic leukemia (3,5).
For the majority of affected males (90%), XLI is
caused by a STS deficiency due to the complete deletion of the STS
gene located on the short arm of the X chromosome (Xp22.3). The
remaining 10% of males demonstrate a point mutation or partial
deletion (4,6,7).
Larger deletions involving neighboring genes (contiguous gene
deletion syndromes) may result in XLI associated with Kallman
syndrome (KS) or X-linked recessive chondrodysplasia punctata
(CDXP) (4,6,7).
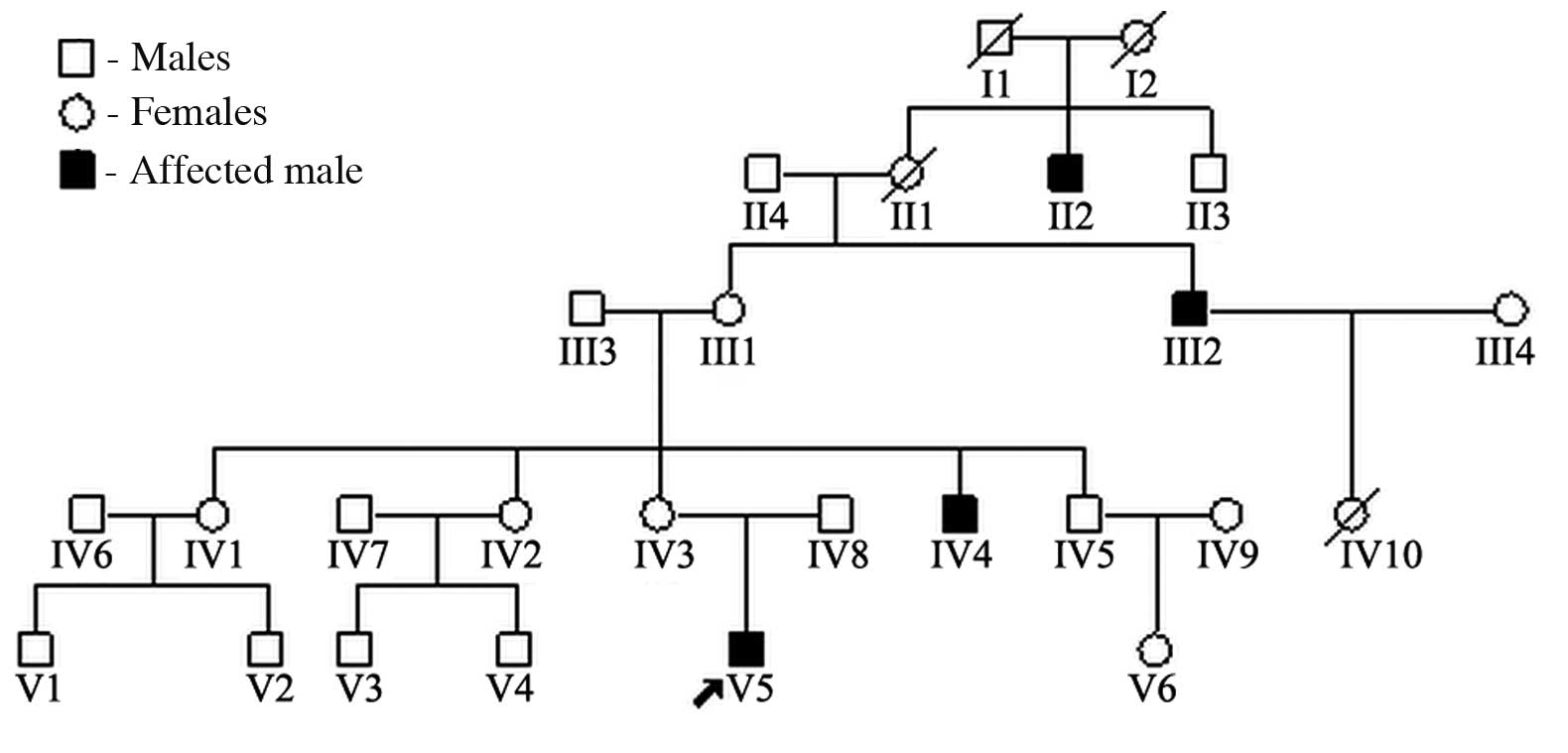
In this study, we report on the case of a
12-year-old male with XLI in association with glomerular sclerosis.
Ichthyosis also affects two other members of the patient’s family.
The case report describes our investigation into the deletion
pattern of the STS gene and flanking regions in DNA samples of the
family members. The study was approved by the Ethics Committee of
The Second Xiangya Hospital of Central South University, Changsha,
Hunan, China and consent was obtained from the the patient’s
family
Case report
Patient history
A 12-year-old male presented with increasing
nocturnal urine volume for five years, pallor for one year and
discontinuous tetany for the previous two days. At seven years old,
the increase in urine volume, ~800 ml/night, in addition to
enuresis were noted, but aggravated gradually. There was no history
of oliguria or edema. The patient was the product of a
non-consanguineous marriage and was delivered by cesarian section
following an uneventful pregnancy. Birth weight was 3050 g and no
neonatal disturbance was recorded. Intelligence and development
were considered normal. When the patient was one year old,
characteristic ichthyosis lesions were noted on the extremities and
abdomen. Similar ichthyotic skin was observed in the maternal uncle
and great uncle (Fig. 1). There
was no family history of renal disease.
Examination
Anthropometrical evaluation revealed that the
patient had a proportionately short stature (SS) with a height of
129 cm (mean height of Chinese 12-year-old male, 150.4±2.6 cm),
below the 3rd percentile (8).
Blood pressure was 120/80 mmHg. Audiometry, visual acuity and
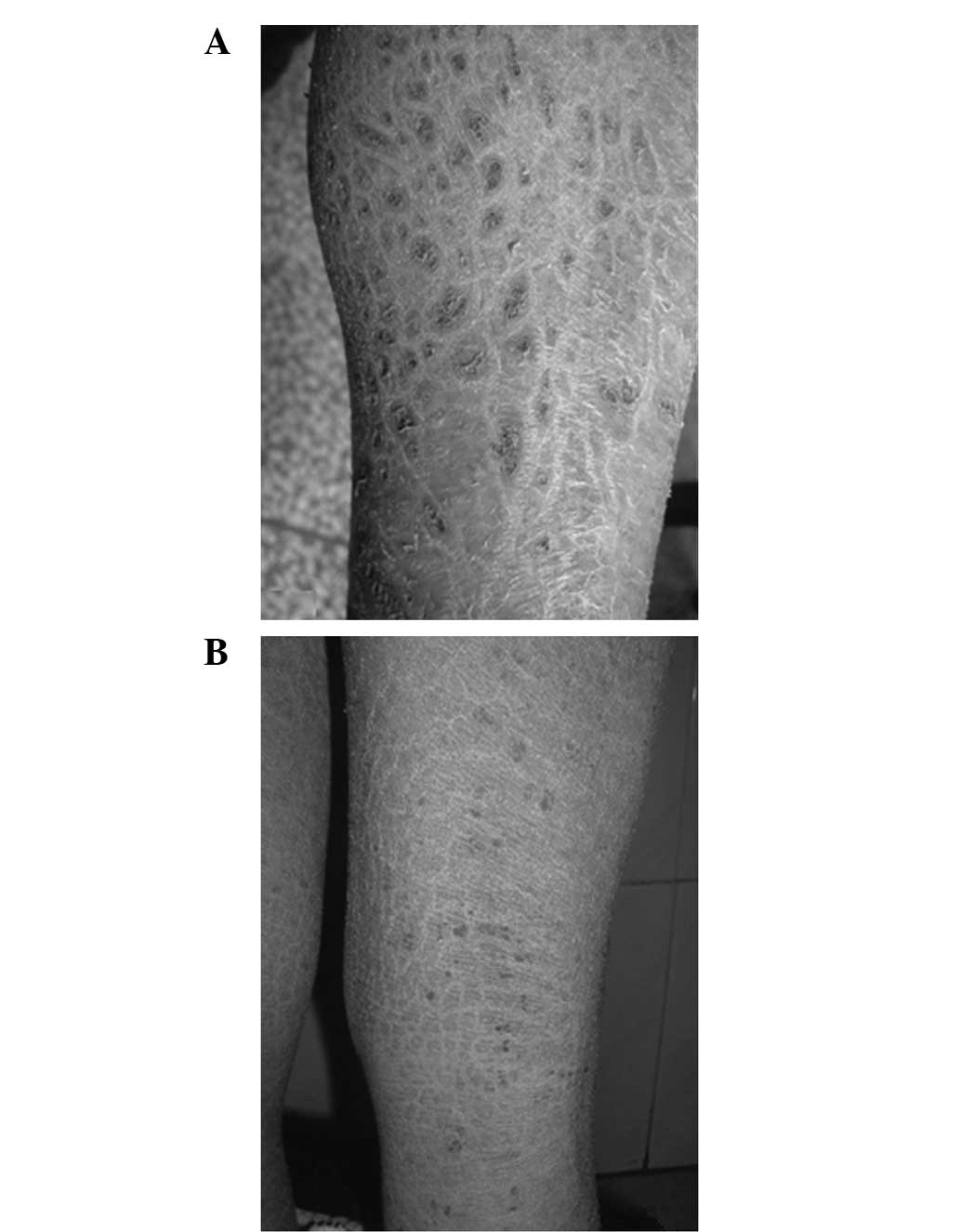
olfactory sensation were normal. Large polygonal dark brown scales
were observed over the extensor and flexor aspects of the lower
limbs (Fig. 2). There were no
features typical of renal osteodystrophy or rickets. Investigation
revealed anemia (Hb 56 g/l), mild proteinuria (0.4 g/day),
hypocalcemia (1.18 mmol/l) and renal failure [blood urea nitrogen
(BUN) 42.53 mmol/l, creatinine (Cr) 696.4 μmol/l]. Serum
cholesterol and serum albumin levels were normal. The serum
phosphate level was higher than normal (2.78 mmol/l). The quantity
of mercury in the urine was normal. Examination of the ocular
fundus and endocrinological studies did not reveal any abnormal
findings. Abdominal ultrasound exhibited small kidneys and no
evidence of reflux nephropathy. The patient was diagnosed with
chronic kidney disease with XLI.
Family members
There were two other male family members affected,
the maternal uncle and great uncle. Physical examination revealed
generalized ichthyosis and the heights of the maternal uncle and
great uncle were 153 cm (average height of individual of the same
age and gender, 169.0±2.7 cm) and 150 cm (average height of
individual of the same age and gender, 166.9±2.8 cm) (9), respectively. Audiometry, visual
acuity and olfactory sensation were normal. Routine laboratory
tests and examination of ocular fundi were normal. The mother did
not exhibit any evidence of ichthyosis and SS. Routine laboratory
tests revealed normal results.
Biopsies
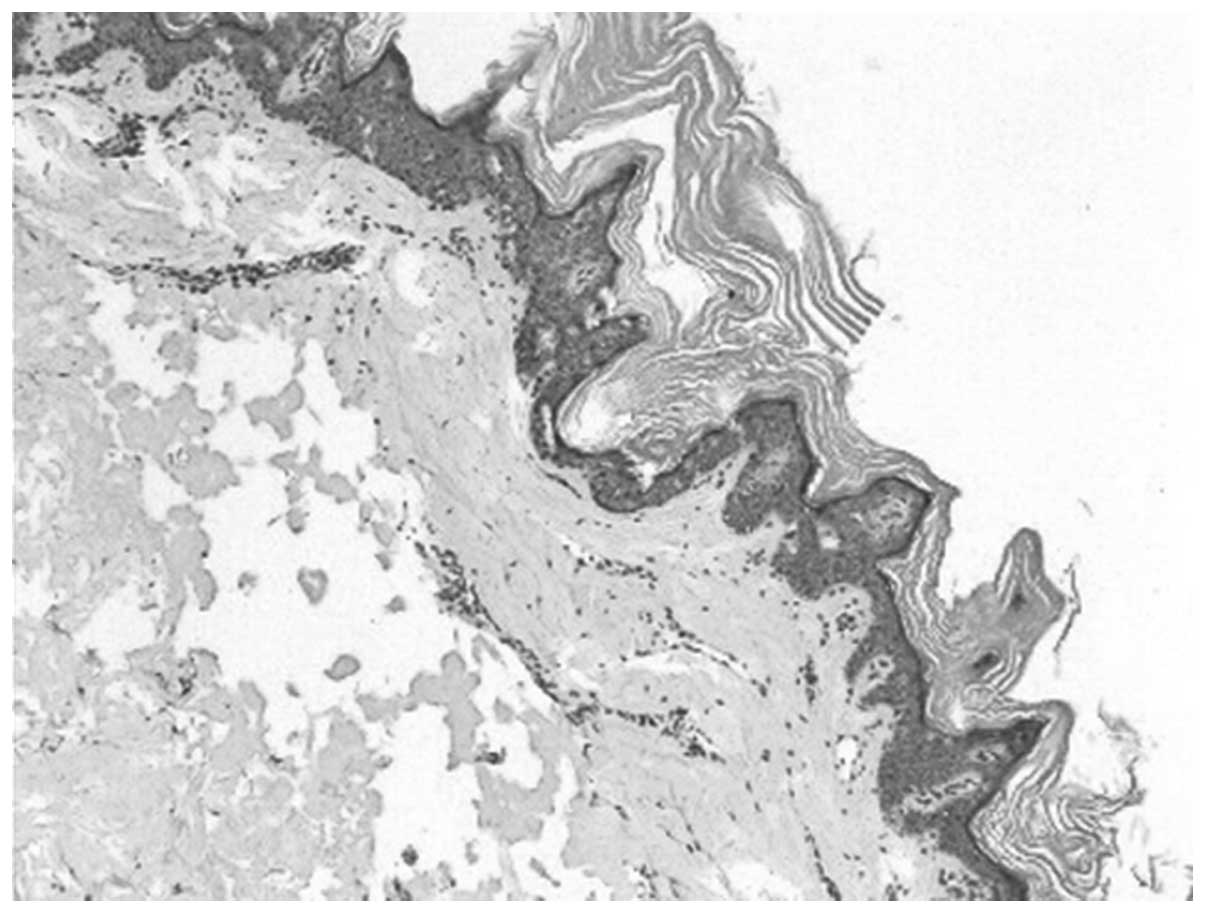
The skin biopsy of the 12-year-old male revealed
slight papillomatous hyperplasia and hyperkeratosis. The granular
layer was normal and the prickle layer was thicker (Fig. 3). A percutaneous renal biopsy was
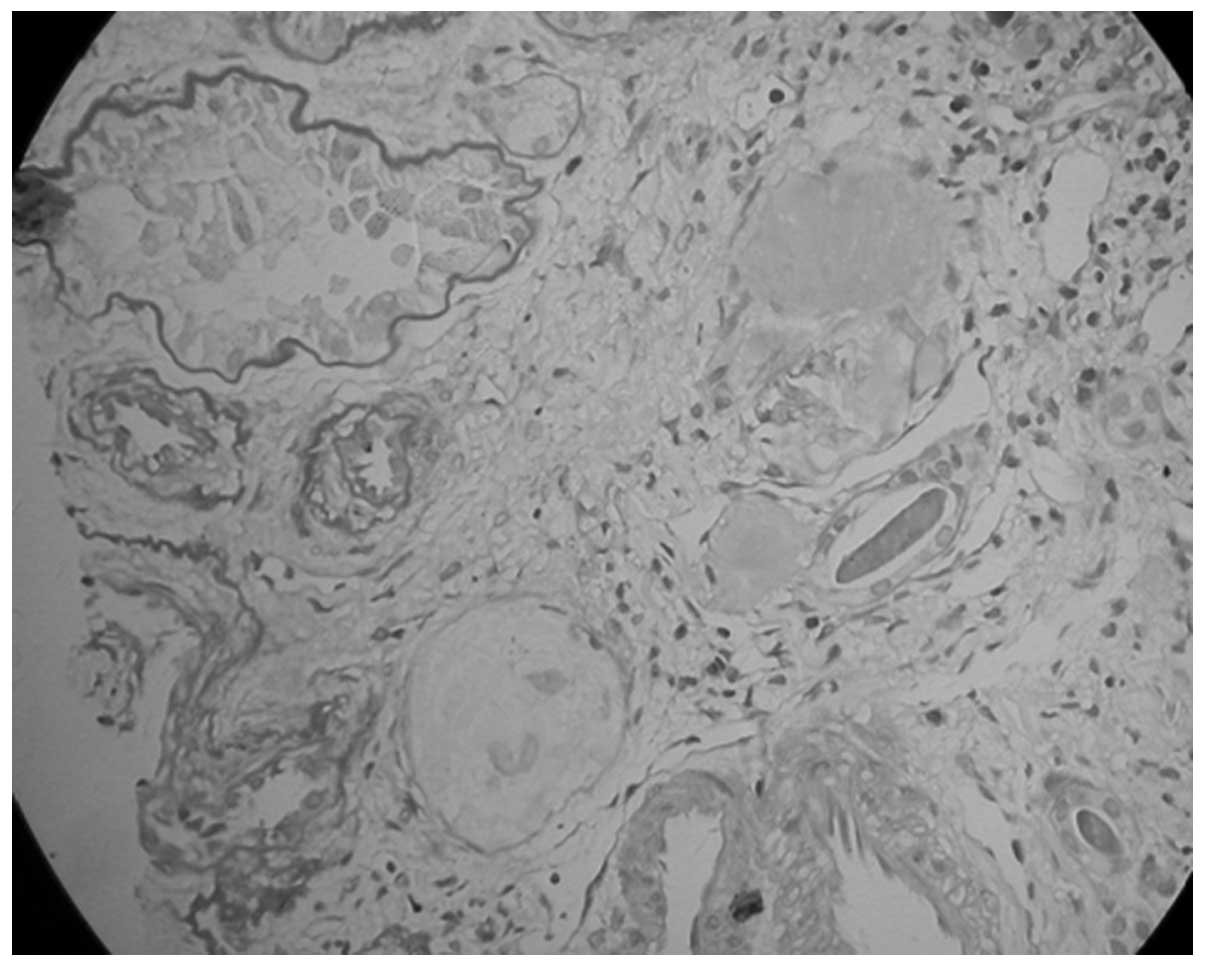
performed. The kidney tissue specimen contained nine glomeruli,
none of which exhibited any evidence of aplasia, hypoplasia or
dysplasia and seven of which were completely obsolescent (Fig. 4). The rest of the glomeruli
revealed mild focal and segmental mesangial hypercellularity
without glomerulosclerosis. The capillary lumina were clear. The
interstitium contained no foam cells or significant vascular
lesions. These demonstrated sclerosing glomerulonephritis and
diffuse interstitial fibrosis. Immunofluorescence of the kidney
biopsy specimen revealed positive mesangial immunostaining for C3
and a weak positive for IgM. Electron microscopy revealed
glomerular sclerosis. There were many filaments in the glomerulus.
The glomerular basement membrane (GBM) was irregularly thick in its
appearance and cloudy. Electron-dense deposits were observed which
exhibited fibrillary glomerulonephritis.
Treatment
The patient started treatment with tertian
hypodermic erythropoietin for anemia, and hemodialysis and
peritoneal dialysis for the management of renal failure. The
patient was discharged whilst on peritoneal dialysis.
Genetic analysis
DNA extraction
Genomic DNA was isolated from the peripheral white
blood cells of the patient, the mother of the patient and the
patient’s maternal uncle and great uncle using the
Puregene® DNA Isolation kit (Gentra, Valencia, CA, USA)
according to the manufacturer’s instructions. Control genomic DNA
was prepared from blood samples of unrelated healthy Chinese
individuals with no evidence of ichthyosis or renal disease.
PCR
The STS gene was analyzed by PCR. The conditions and
primers used to amplify the sequence
telomeric-DXS89-DXS996-DXS1139-DXS1130-5′STS-3′STSDXS1131-DXS1133-DXS237-DXS1132-DXF22S1-DXS278-K
AL-DXS1134-centromeric of the STS gene are as previously described
(10–12). All procedures were repeated three
times.
Results
PCR
Results of the PCR are shown in Fig. 5 and Table I. In the patient and the patient’s
maternal uncle and great uncle, DXS89, DXS996, DXS278, KAL and
DXS1134 were present, whereas DXS1139, DXS1130, 5′STS, 3′STS,
DXS1131, DXS1133, DXS237, DXS1132 and DXF22S1 failed to amplify,
suggesting a large deletion involving the region from DXS1139 to
DXF22S1. It could not be confirmed whether there was a difference
between the three afffected male family members with regard to
DXS89, DXS996, DXS278, KAL and DXS1134 as the PCR results were
qualitative. The mother, who was assumed to be a carrier, exhibited
a normal amplifying pattern in these loci.
 | Figure 5PCR amplification of the STS gene and
flanking regions. 1, normal control; 2, mother; 3, maternal uncle;
4, 12-year-old male patient; 5, great uncle. All the genes were
amplified in the mother. DXS89, DXS996, DXS278 and DXS1134 were
amplified in the patient and the patient’s maternal uncle and great
uncle, whereas DXS1139, DXS1130, 5′STS, 3′STS, DXS1133, DXS237 and
DXF22S1 were not amplified. STS, steroid sulfatase. |
 | Table IDeletion map of the family. |
Table I
Deletion map of the family.
|
Gene |
|---|
|
|
|---|
| No. | DXS89 | DXS996 | DXS1139 | DXS1130 | 5′STS | 3′STS | DXS1133 | DXS237 | DXF22S1 | DXS278 | DXS1134 |
|---|
| 1 | + | + | + | + | + | + | + | + | + | + | + |
| 2 | + | + | + | + | + | + | + | + | + | + | + |
| 3 | + | + | − | − | − | − | − | − | − | + | + |
| 4 | + | + | − | − | − | − | − | − | − | + | + |
| 5 | + | + | − | − | − | − | − | − | − | + | + |
Discussion
Congenital ichthyosis may be associated with a
variety of other disorders, including renal disease (13–15).
In most instances, it takes the form of nonbullous congenital
ichthyosiform erythroderma, which is subject to autosomal recessive
inheritance and is assumed to be caused by mutations in the TGM1
gene, located on chromosome 14 (16). Martul et al(17) described the concurrence of XLI and
renal malformation. Matsukura et al(18) reported an 8-year-old male who
presented with steroid-resistant nephrotic syndrome (SRNS)
associated with XLI. The findings of the kidney biopsy were
compatible with minimal change disease (MCD). Despite
immunosuppressants and prednisolone, no clinical response was
achieved. The 8-year-old patient rapidly reached end-stage renal
failure and underwent renal transplantation. Krishnamurthy et
al(19) described a
10-year-old male with XLI, KS and unilateral renal agenesis who
presented with nephrotic syndrome. The 10-year-old patient started
treatment with prednisolone tablets at 2 mg/kg/day and went into
remission after six days. Mishra et al(20) described a four and a half-year-old
male who presented with SRNS with an underlying ichthyotic skin
disorder present since birth. Renal biopsy revealed MCD. Genetic
analysis was performed on male family members with similar skin
manifestations, which revealed the deletion of the STS gene
spanning the 3′ and 5′ terminals. The patient was started on a
cyclosporine regimen and remission was achieved in five weeks. All
patients in the latter three cases presented with nephrotic
syndrome, and the second and third patients presented with
cryptorchidism, both of which were absent in the 12-year-old male
subject of our study. In this study, the presence of ichthyosis in
a maternal uncle and great uncle suggested that the disorder is
X-linked and DNA analysis of the STS gene on chromosome Xp22.32
confirmed this.
Of the XLI patients, ~90% have large deletions of
the STS gene and its flanking sequences. Such deletions may
occasionally extend to involve neighboring genes, caused by a
contiguous gene defect. Therefore, XLI may be associated with KS,
mental retardation, CDXP and SS. In our study, the patient and the
patient’s affected male relatives demonstrated neither hypogonadism
nor anosmia. However, the patient and the patient’s maternal uncle
and great uncle all displayed SS. By analyzing the DNA samples of
the family, we found that the gene deletion did not extend to the
causative gene of KS (KAL gene) and SS (SHOX gene; data not shown).
In our study, PCR analysis on the patients revealed an interstitial
deletion with the telomeric breakpoint located between DXS996 and
DXS1139, and the centromeric breakpoint located between DXF22S1 and
DXS278. Ballabio et al(21), Saeki et al(22), Jimenez Vaca et al(23) and Aviram-Goldring et
al(24) demonstrated the
molecular heterogeneity of the STS deficiency in their studies of
XLI patients of various races. Their results revealed heterogeneity
in the deletion pattern among all patients with XLI. The
breakpoints involved loci from DXS1139 to DXS278, the flanking
sequences of the STS gene. The most common deletion pattern is a
complete deletion involving the entire region from DXS1139 to
DXF22S1. The patients in this study had the same deletion.
STS is a membrane-bound microsomal enzyme,
ubiquitously expressed in mammalian tissues, that hydrolyzes
various 3 β-hydroxysteroid sulfates (4,25).
The enzyme is crucial in the conversion of sulfated steroid
precursors to estrogens during human pregnancies and in the
desulfation of cholesterol sulfate, an important step in skin
metabolism. STS deficiency in XLI leads to cholesterol sulfate
accumulation, which induces transglutaminase-1 (TGM-1) dysfunction.
Cholesterol sulfate accumulates in the stratum corneum, producing a
barrier abnormality in intact skin, extracellular anomalies in the
isolated stratum corneum and inhibiting the formation of
detergent-insoluble membrane domains (25). Cholesterol sulfate interferes with
the normal activity of TGM-1, it is this deficiency that causes
lamellar ichthyosis (26). It has
been proposed that XLI is a consequence of cholesterol sulfate
induced TGM-1 dysfunction (27).
TGM-1 is expressed in large quantities in skin, lung, liver and
kidney epithelial tissues. The formation of covalently cross-linked
multimolecular complexes by TGM-1 is an important mechanism for
maintaining the structural integrity of simple epithelial cells,
particularly at cadherin-based adherens junctions between the
epithelial cells of the lung, liver and kidney (28). Due to the fact that the slit
diaphragm of the glomerular visceral epithelial cell is a modified
adherens junction (29), it has
been postulated that cholesterol sulfate accumulation may interfere
with normal slit diaphragm function, resulting in proteinuria
(29). However, XLI associated
with renal disease is uncommon and further studies are required in
order to determine whether the STS gene or the co-deleted flanking
sequences are causative of the renal disease observed in XLI.
Acknowledgements
We are grateful to all members of the XLI family for
participating in this study.
References
|
1
|
Webster D, France JT, Shapiro LJ and Weiss
R: X-linked ichthyosis due to steroid sulphatase deficiency.
Lancet. 1:70–72. 1978.
|
|
2
|
Paige DG, Emilion GG, Bouloux PM and
Harper JI: A clinical and genetic study of X-linked recessive
ichthyosis and contiguous gene defects. Br J Dermatol. 131:622–629.
1994. View Article : Google Scholar
|
|
3
|
Gohlke BC, Haug K, Fukami M, et al:
Interstitial deletion in Xp22.3 is associated with X linked
ichthyosis, mental retardation, and epilepsy. J Med Genet.
37:600–602. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hernández-Martin A, González-Sarmiento R
and De Unamuno P: X-linked ichthyosis: an update. Br J Dermatol.
141:617–627. 1999.
|
|
5
|
Richard G: Molecular genetics of the
ichthyoses. Am J Med Genet C Semin Med Genet. 131C:32–44. 2004.
View Article : Google Scholar
|
|
6
|
Ballabio A, Sebastio G, Carrozzo R, et al:
Deletions of the steroid sulphatase gene in ‘classical’ X-linked
ichthyosis and in X-linked ichthyosis associated with Kallmann
syndrome. Hum Genet. 77:338–341. 1987.
|
|
7
|
Bonifas JM, Morley BJ, Oakey RE, Kan YW
and Epstein EJ Jr: Cloning of a cDNA for steroid sulfatase:
Frequent occurrence of gene deletions in patients with recessive X
chromosome-linked ichthyosis. Proc Natl Acad Sci USA. 84:9248–9251.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu YM, Jiang ZF and Zhu FT: Practical
Paediatrics. 7th edition. People’s Medical Publishing House;
Beijing: pp. 23–31. 2002
|
|
9
|
Yang XG, Li YP, Ma GS, et al: Study on
weight and height of the Chinese people and the differences between
1992 and 2002. Zhonghua Liu Xing Bing Xue Za Zhi. 26:489–493.
2005.(In Chinese).
|
|
10
|
Ballabio A, Ranier JE, Chamberlain JS,
Zollo M and Caskey CT: Screening for steroid sulfatase (STS) gene
deletion by multiplex DNA amplification. Hum Genet. 84:571–573.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schaefer L, Ferrero GB, Grillo A, et al: A
high resolution deletion map of human chromosome Xp22. Nat Genet.
4:272–279. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Valdes-Flores M, Kofman-Alfaro SH, Jimenez
Vaca AL and Cuevas-Covarrubias SA: Mutation report: a novel partial
deletion of exons 2–10 of the STS gene in recessive X-linked
ichthyosis. J Invest Dermatol. 114:591–593. 2000.
|
|
13
|
Goyer RA, Reynolds J Jr, Burke J and
Burkholder P: Hereditary renal disease with neurosensory hearing
loss, prolinuria and ichthyosis. Am J Med Sci. 256:166–179. 1968.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rayner A, Lampert RP and Rennert OM:
Familial ichthyosis, dwarfism, mental retardation, and renal
disease. J Pediatr. 92:766–768. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deal JE, Barratt TM and Dillon MJ: Fanconi
syndrome, ichthyosis, dysmorphism, jaundice and diarrhoea - a new
syndrome. Pediatr Nephrol. 4:308–313. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laiho E, Ignatius J, Mikkola H, et al:
Transglutaminase 1 mutations in autosomal recessive congenital
ichthyosis: private and recurrent mutations in an isolated
population. Am J Hum Genet. 61:529–538. 1997. View Article : Google Scholar
|
|
17
|
Martul P, Pineda J, Levilliers J, et al:
Hypogonadotrophic hypogonadism with hyposmia, X-linked ichthyosis,
and renal malformation syndrome. J Clin Endocrinol (Oxf).
42:121–128. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsukura H, Fuchizawa T, Ohtsuki A, et
al: End-stage renal failure in a child with X-linked ichthyosis.
Pediatr Nephrol. 18:297–300. 2003.PubMed/NCBI
|
|
19
|
Krishnamurthy S, Kapoor S and Yadav S:
Nephrotic syndrome with X-linked ichthyosis, Kallmann Syndrome and
unilateral renal agenesis. Indian Pediatr. 44:301–303.
2007.PubMed/NCBI
|
|
20
|
Mishra K, Batra VV, Basu S, et al:
Steroid-resistant nephrotic syndrome associated with steroid
sulfatase deficiency-x-linked recessive ichthyosis: a case report
and review of literature. Eur J Pediatr. 171:847–850. 2012.
View Article : Google Scholar
|
|
21
|
Ballabio A, Carrozzo R, Parenti G, et al:
Molecular heterogeneity of steroid sulfatase deficiency: A
multicenter study on 57 unrelated patients at DNA and protein
levels. Genomics. 4:36–40. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saeki H, Kuwata S, Nakagawa H, Shimada S,
Tamaki K and Ishibashi Y: Deletion pattern of steroid sulphatase
gene in Japanese patients with X-linked ichthyosis. Br J Dermatol.
139:96–98. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jimenez Vaca AL, Valdes-Flores Mdel R,
Rivera-Vega MR, González-Huerta LM, Kofman-Alfaro SH and
Cuevas-Covarrubias SA: Deletion pattern of the STS gene in X-linked
ichthyosis in a Mexican population. Mol Med. 7:845–849.
2001.PubMed/NCBI
|
|
24
|
Aviram-Goldring A, Goldman B,
Netanelov-Shapira I, et al: Deletion patterns of the STS gene and
flanking sequences in Israeli X-linked ichthyosis patients and
carriers: analysis by polymerase chain reaction and fluorescence in
situ hybridization techniques. Int J Dermatol. 39:182–187. 2000.
View Article : Google Scholar
|
|
25
|
Valdes-Flores M, Kofman-Alfaro SH, Vaca AL
and Cuevas-Covarrubias SA: Deletion of exons 1–5 of the STS gene
causing X-linked ichthyosis. J Invest Dermatol. 116:456–458.
2001.
|
|
26
|
Russell L, DiGiovanna J, Rogers G, et al:
Mutations in the gene for transglutaminase 1 in autosomal recessive
lamellar ichthyosis. Nat Genet. 9:279–283. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nemes Z, Demény M, Marekov LN, Fésüs L and
Steinert PM: Cholesterol 3-sulfate interferes with cornified
envelope assembly by diverting transglutaminase 1 activity from the
formation of cross-links and esters to the hydrolysis of glutamine.
J Biol Chem. 275:2636–2646. 2000. View Article : Google Scholar
|
|
28
|
Hiiragi T, Sasaki H, Nagafuchi A, et al:
Transglutaminase type 1 and its cross-linking activity are
concentrated at adherens junctions in simple epithelial cells. J
Biol Chem. 274:34148–34154. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reiser J, Kriz W, Kretzler M and Mundel P:
The glomerular slit diaphragm is a modified adherens junction. J Am
Soc Nephrol. 11:1–8. 2000.PubMed/NCBI
|