Introduction
Intracerebral hemorrhage (ICH) constitutes 15% of
all strokes in the USA and Europe, and 20–30% in Asian and African
populations (1). Approximately
30–40% of ischemic strokes undergo a certain degree of hemorrhagic
conversion (2). ICH is associated
with high mortality and disability. To date, surgical approaches
for the treatment of ICH have not been particularly effective and
no satisfactory drug treatment is currently used in clinical
practice (3,4).
ICH is a dynamic, serial process in which early
hematoma expansion with excitotoxicity, perihematomal inflammation
and apoptosis occurs (5).
Consequently, prospective treatments reducing brain edema or
apoptotic cell death may provide a neuroprotective strategy for ICH
patients.
Erythropoietin (EPO) is a glycoprotein that is
predominantly produced in the adult kidneys and the liver. EPO and
the EPO receptor (EPOR) are essential for erythropoiesis and are
also expressed in neurons, astrocytes and cerebral endothelial
cells in the brain. EPO has been shown to exhibit anti-apoptotic,
anti-inflammatory, anti-oxidative, angiogenetic and neurotrophic
properties (6). Its potential
therapeutic effect has been demonstrated in the animal model of
cerebral ischemia (7–9), spinal cord injury (10,11),
traumatic brain injury (12,13)
and subarachnoid hemorrhage (14–16).
Recently, studies have demonstrated the beneficial
involvement of EPO in ICH, possibly through its anti-inflammatory
and anti-apoptotic effects (17–20).
However, the molecular and cellular mechanisms and the signaling
pathway underlying these effects remains to be elucidated.
The present study aimed to investigate the effect of
rhEPO in an autologous blood-injected rat model of ICH and the
underlying molecular and cellular mechanisms, particularly the
involvement of the PI3K signaling pathway.
Materials and methods
Animal preparation and ICH model
The rats were obtained from the Experimental Animal
Center of the 3rd Military Medical University (Chongqing, China)
and the animal protocol for this study was approved by the
Institutional Animal Care and Use Committee of the Chongqing
University of Medical Sciences (Chonqing, China). Adult male
Sprague-Dawley rats (n=64; weight, 280–300 g) were used in this
study. Animals were allowed access to food and water ad
libitum prior to the experiments. The ICH model was performed
as described previously (21). The
rats were anesthetized with an intraperitoneal injection of 10%
chloral hydrate at 400 mg/kg and positioned in a stereotaxic frame
(David Kopf Instruments, Tujunga, CA, USA). Under sterile
conditions, a cranial burr hole (1 mm) was drilled near the right
coronal suture, 3.5 mm lateral to the midline. A 26-gauge needle
was inserted stereotaxically into the right basal ganglia
(coordinates: 0.2 mm anterior, 5.5 mm ventral and 3.5 mm lateral to
the bregma). Autologous whole blood (100 μl, with no
anticoagulants) was injected at a speed of 10 μl/min with a
microinfusion pump (Braun, Melsungen, Germany). The needle was
removed and the skin incision was sutured following infusion. All
rats were allowed to recover fully under observation. Sham-operated
animals were subjected to needle insertion only.
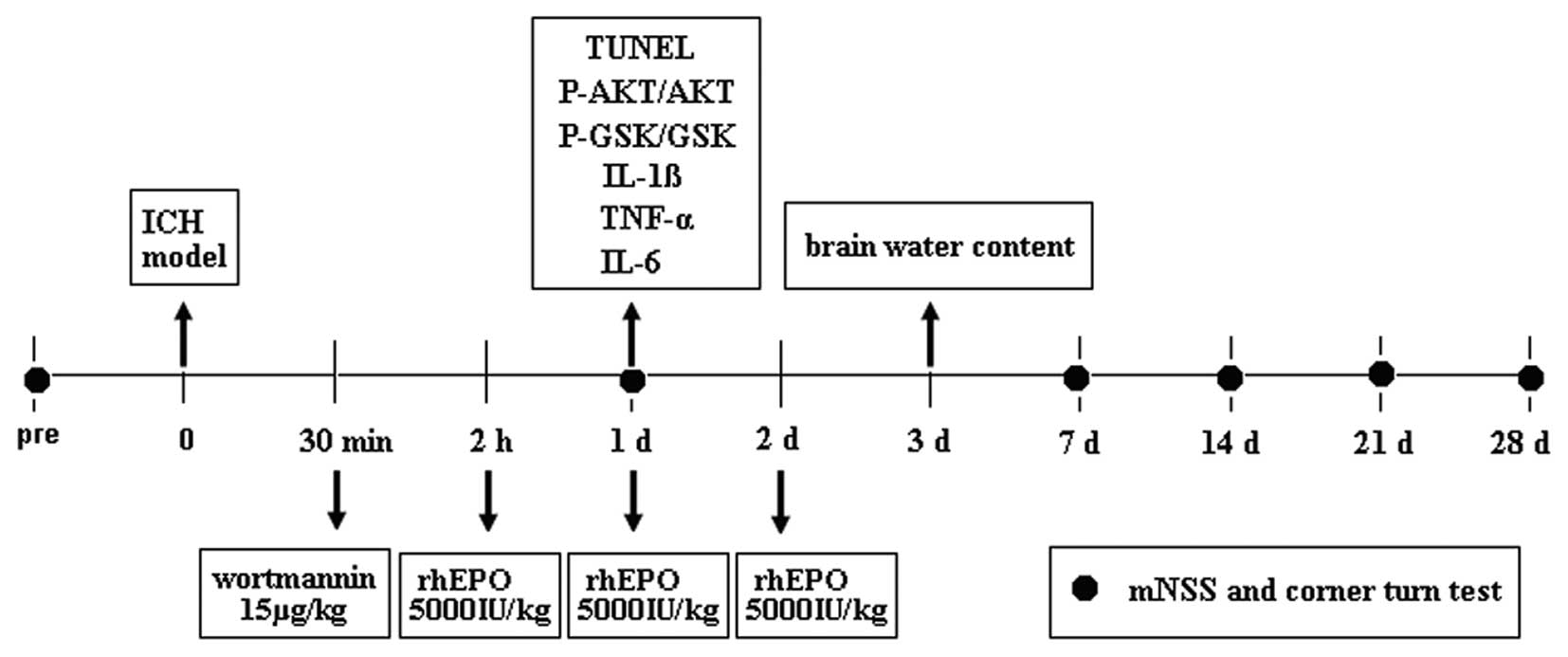
Experimental groups and pharmacological
interventions
The rats were divided into the following four
groups: Group 1, sham (n=18); group 2, ICH + vehicle (n=18); group
3, ICH + rhEPO (n=18); and group 4, ICH + rhEPO + wortmannin
(n=18). rhEPO (H. Lundbeck A/S, Copenhagen, Denmark) was
administered intraperitoneally at 2, 24 and 48 h at 5000 IU/kg body
weight following ICH. The experimental design is shown in Fig. 1. The animals in the ICH + vehicle
group received vehicle intraperitoneally at 2, 24 and 48 h. The ICH
rats received the PI3K inhibitor wortmannin (15 μg/kg)
intravenously 90 min prior to rhEPO injection. The vehicle-treated
animals received normal saline at a volume equivalent to that
administered to animals in the intervention groups. All chemicals
were purchased from Sigma-Aldrich (St. Louis, MO, USA) and
processed according to previously published protocols (22,23).
The dosage of rhEPO was based on previous studies on traumatic
brain injury and ischemic stroke (19,12).
Assessment of behavioral outcome
Behavioral outcomes were measured using the modified
neurological severity score (mNSS) (24) and the corner turn test score
(25). These were performed by an
observer who was blinded to the individual treatment status of the
animals. Behavioral outcomes for each rat were tested 1, 7, 14, 21
and 28 days following ICH. The mNSS is a composite of motor (muscle
status and abnormal movement), sensory (visual, tactile and
proprioceptive), reflex and balance tests. Neurological function
was graded on a scale of 0 to 18 (normal score, 0; maximal deficit
score, 18), where higher scores indicate more severe behavioral
deficits. The corner test detects impairment of sensorimotor
function. Briefly, one rat was placed between two boards that were
attached with the edges at a 30° angle to each other. The control
rat turned either left or right, but the hemorrhagic rat
preferentially turned toward the non-impaired side. The number of
right turns was recorded from 10 tests for each rat.
Measurement of brain water content
The brain water content was measured 3 days
following ICH injury. The rats were decapitated under deep
anesthesia and the brains were immediately removed and cut into
4-mm sections. Each section was divided into four parts;
ipsilateral and contralateral basal ganglia, and ipsilateral and
contralateral cortex. The cerebellum was collected as the internal
control. Tissue samples were weighed on an electronic analytical
balance (Changzhou Instruments, Changzhou, China) to the nearest
0.1 mg to obtain the wet weight. The brain samples were then dried
at 100°C in an electric blast-drying oven (Sida Apparatus, Jiangsu,
China) for 24 h to obtain the dry weight. The brain water content
(%) was calculated as (wet weight - dry weight)/wet weight ×
100.
Determination of inflammatory cytokines
in hemorrhagic brain tissue
To determine the levels of inflammatory cytokines in
the striatal tissue, ipsilateral striatal tissues were collected 24
h following ICH. The tissues were lysed (40%, wt/vol) with 0.01
mol/l phosphate-buffered saline (pH 7.4) containing a protease
inhibitor cocktail (Roche, Indianapolis, IN, USA) and homogenized.
The homogenates were then centrifuged at 7500 × g for 20 min at 4°C
using a high-speed cryostat centrifuge (3K30; Sigma Centrifuges
GmbH, Osterode am Harz, Germany). The levels of tumor necrosis
factor-α (TNF-α), interleukin (IL)-1β and IL-6 in the supernatant
were determined by enzyme-linked immunosorbent assay (ELISA) using
commercial kits (DuoSetELISA Development Systems; R&D Systems,
Minneapolis, MN, USA) according to the manufacturer’s instructions
via an ELISA analyzer (Coda, Bio-Rad, Hercules, CA, USA).
Immunofluorescent staining
The rats were euthanized 24 h following surgery and
brain specimens were processed for immunostaining as previously
described (1). Double
immunofluorescence staining was performed using the neuronal marker
anti-NeuN monoAb (1:100, Millipore, Temecula, CA, USA) and terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; Roche).
Once stained, the specimens were analyzed under a fluorescence
microscope (Olympus BX51; Olympus, Melville, NY, USA). The
TUNEL-positive cells were counted at the center of the hemorrhagic
lesion. Three perihematomal regions of the ipsilateral cerebral
hemisphere were used for cell counting. The result was presented as
the cell density of TUNEL-positive or TUNEL/NeuN-positive cells.
Four rats per group were used for intergroup comparisons.
Western blot analysis
Rats were sacrificed 24 h after ICH and western blot
analysis was performed as previously described (26). The brain samples were homogenized
and the total proteins were extracted using radio
immunoprecipitation assay lysis buffer (Beyotime Biotech. Co.,
Nanjing, China). Samples with an equal quantity of protein (50 μg)
were separated on 10% sodium dodecyl sulfate polyacrylamide gels,
transferred onto nitrocellulose membranes and blocked in 5% non-fat
dry milk buffer at 4°C overnight. The membranes were then incubated
overnight at 4°C with monoclonal rabbit anti-phospho-glycogen
synthase kinase (GSK)-3β (Ser9; 1:1,000; Cell Signaling Technology,
Inc., Beverly, MA, USA), monoclonal rabbit anti-total GSK-3β
(1:2,000; Cell Signaling Technology, Inc.), polyclonal rabbit
anti-phospho-Akt (Ser473; 1:1000; Cell Signaling Technology, Inc.)
and polyclonal rabbit anti-Akt (1:1,000; Cell Signaling Technology,
Inc.) for 1 h, incubated with Enhanced Chemiluminescence Plus
solution (GE Healthcare, Little Chalfont, UK) for 5 min and were
densitometrically quantified and analyzed using an NIH image
analysis system. Phosphorylation levels of protein were indicated
by the ratio of phospho-protein to total protein. The value was
then normalized to the mean value of the sham-operated group for
comparison.
Statistical analysis
The data are expressed as the mean ± SEM and were
statistically analyzed using one-way analysis of variance (ANOVA)
followed by Tukey’s post hoc test. All behavioral data are
expressed as the mean ± SEM of sham performance and analyzed with
Kruskal-Wallis one-way ANOVA on ranks, followed by the
Student-Newman-Keuls method. P<0.05 was considered indicate a
statistically significant difference. All statistical analyses were
performed using SigmaPlot version 10.0 (Systat Software, Inc., San
Jose, CA, USA) for Windows.
Results
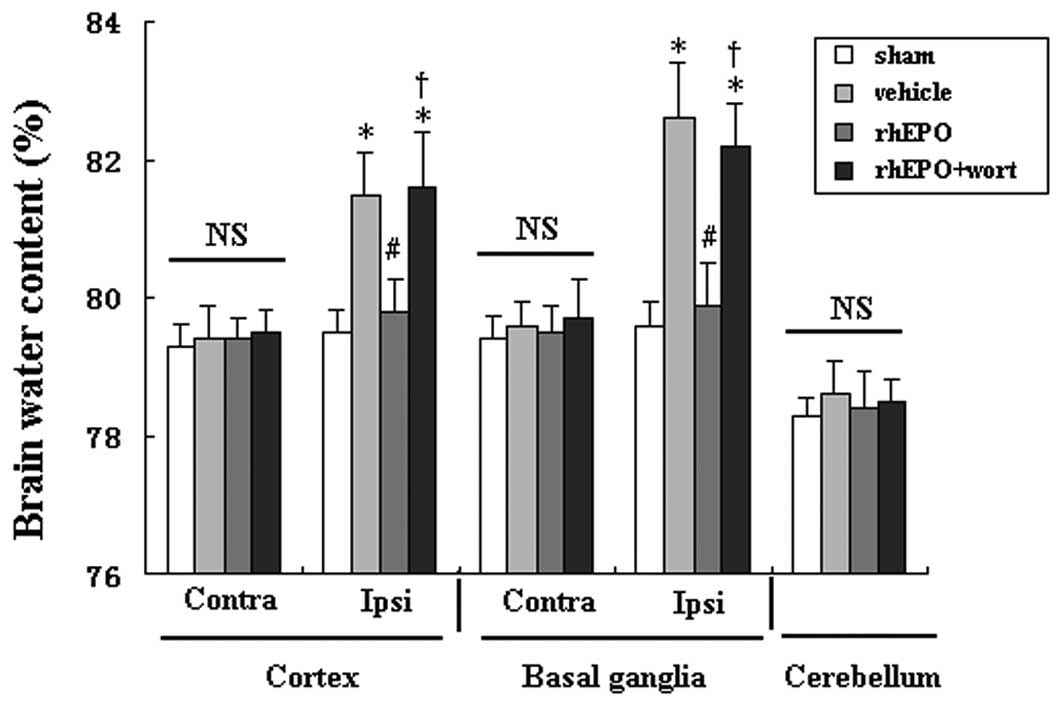
rhEPO attenuates brain edema 72 h
following ICH
Brain edema was evaluated 3 days following ICH (n=6
per group). Rats with intracerebral hemorrhage showed significant
brain edema in the ipislateral tissue compared with the sham group
(P<0.05). rhEPO treatment significantly reduced brain water
content in the ipsilateral basal ganglia and cortex in hemorrhagic
rats compared with rats treated with vehicle (P<0.05, Fig. 2). However, administration of the
PI3K inhibitor wortmannin (15 μg/kg) prior to rhEPO treatment
completely abolished its effect (P<0.05 compared with rhEPO)
without exacerbating brain edema in hemorrhagic rats. No
significant differences were identified in the contralateral
cortex, contralateral basal ganglia or cerebellum among all
groups.
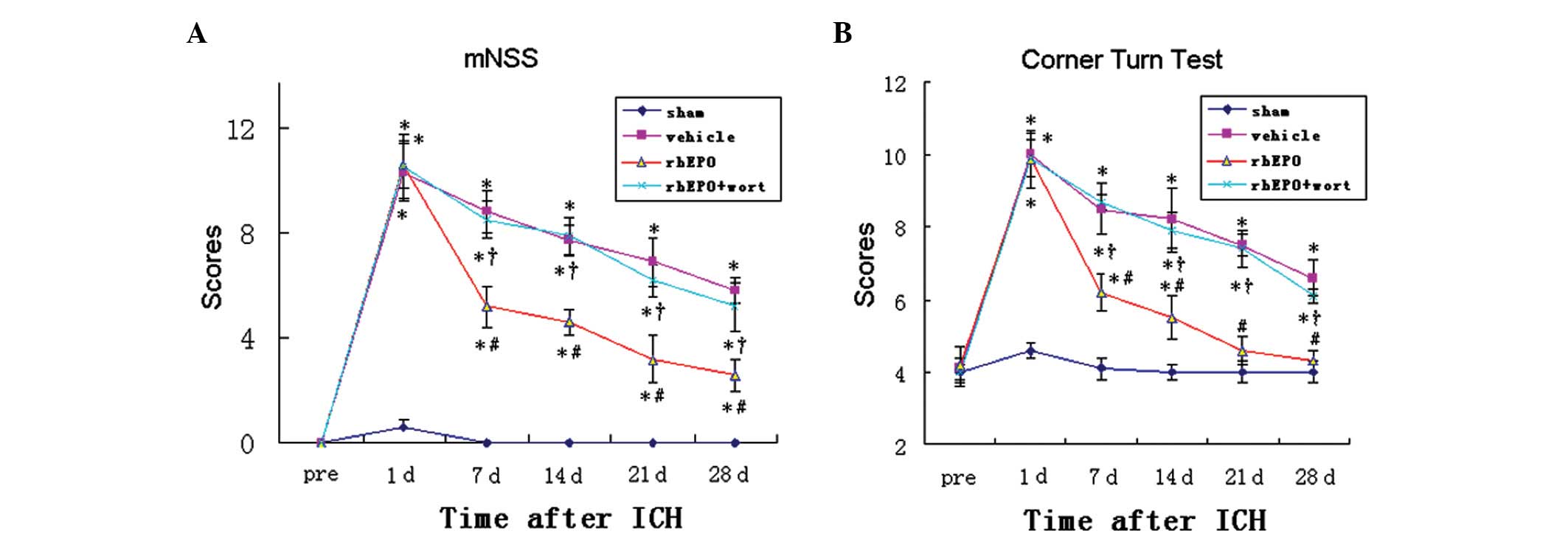
rhEPO attenuates behavioral deficits at
days 7 to 28 following ICH
Rats from the sham group showed no neurological
deficits at any time-point. Day 1 post-ICH, all rats with ICH
showed significantly increased scores in mNSS (P<0.05, Fig. 3A) and the corner turn test
(P<0.05, Fig. 3B), suggesting
that ICH resulted in marked behavioral deficits. The scores of the
tests gradually decreased from day 7 to 28 following ICH. Compared
with the vehicle-treated group, the rhEPO group showed
significantly lower scores in the tests at days 7, 14, 21 and 28
after ICH, suggesting that rhEPO treatment significantly improved
the neurological function in these rats from day 7 onwards. To
determine whether the rhEPO-mediated behavioral improvements were
associated with activation of the PI3K-Akt signaling pathway, the
PI3K inhibitor wortmannin was injected prior to the administration
of rhEPO. The group receiving co-administration of rhEPO and
wortmannin showed significantly higher scores in the two tests at
all time-points from day 7 to 28 compared with the group with rhEPO
alone. Moreover, no difference was identified between the
vehicle-treated group and the rhEPO + wortmannin co-treated group
at all time-points, suggesting that inhibiting the activation of
the PI3K-Akt signaling pathway abolished the beneficial effect of
rhEPO on behavioral outcomes.
rhEPO reduces levels of proinflammatory
cytokines in striatal tissue
Proinflammatory cytokine (IL-1β, TNF-α, and IL-6)
levels in the ipsilateral striatum were determined by ELISA at day
1 following ICH. Compared with the sham-operated rats,
vehicle-treated hemorrhagic rats exhibited significantly higher
levels of IL-1β, TNF-α and IL-6 (Fig.
4A–C, P<0.05), suggesting an ICH-induced inflammatory
response. Treatment with rhEPO significantly reversed the
ICH-induced upregulation of IL-1β TNF-α and IL-6, reaching that of
the sham-operated group. However, wortmannin treatment inhibited
the effect of rhEPO on attenuating the inflammatory response. This
suggested that the activation of the PI3K-Akt signaling pathway is
involved in the rhEPO-mediated inhibition of the inflammatory
response following ICH.
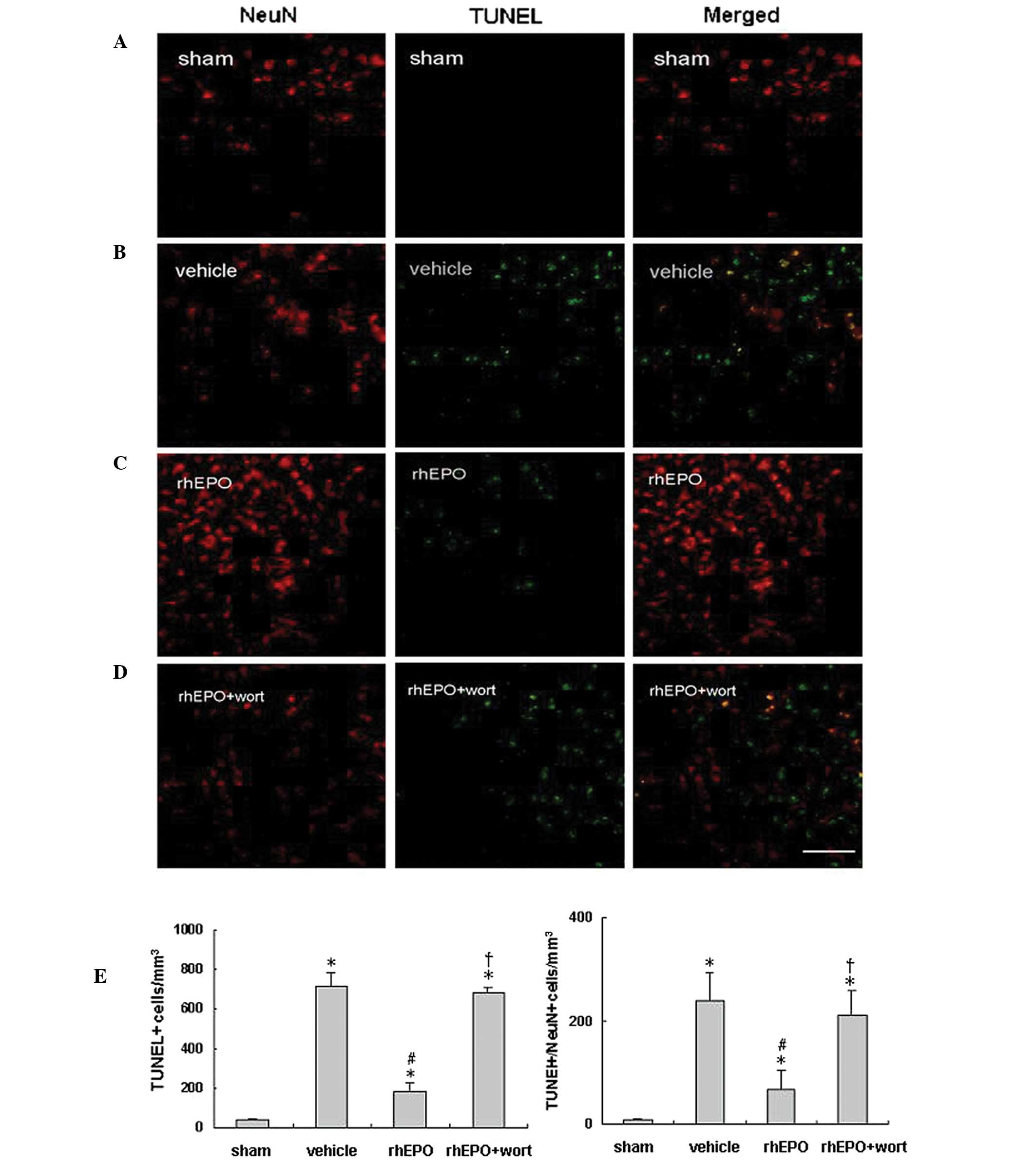
rhEPO reduces neuronal apoptosis 24 h
following ICH
Neuronal apoptosis was analyzed in rats from each
group (n=6 per group) 24 h following ICH or sham operation.
Immunoreactivity of NeuN (red) and TUNEL staining (green) were used
to identify neuronal apoptosis in the perihematomal area or near
the needle tract in the sham animals. Representative images for
each group are shown in Fig. 5.
Compared with the sham-operated group (Fig. 5A), the vehicle-treated hemorrhagic
group (Fig. 5B) showed a decrease
in NeuN-positive cells, but significant increases in TUNEL-positive
and TUNEL/NeuN double-positive cells (Fig. 5E) in the perihematomal area,
suggesting ICH-induced apoptosis and neuronal loss in the injured
area. rhEPO treatment restored NeuN-positive cells and decreased
neuronal apoptosis (Fig. 5C), as
shown with significantly decreased TUNEL- and TUNEL/NeuN
double-positive cells compared with the vehicle-treated group
(Fig. 5E, P<0.05).
Administration of wortmannin prior to rhEPO treatment abolished the
anti-apoptotic effect of rhEPO (Fig.
5C and D; P<0.05 vs. the rhEPO-treated group), and no
significant difference was observed between the vehicle- and rhEPO
+ wortmannin-treated groups (P>0.05).
rhEPO activates the PI3K-Akt signaling
pathway and decreases p-GSK-3β expression 24 h after ICH
To further study the involvement of the PI3K-Akt
signaling pathway in the rhEPO-mediated neuroprotective and
anti-apoptotic effects, western blot analysis was conducted. The
level of phosphorylated Akt (p-Akt, Ser473) and GSK-3β (p-GSK-3β,
Tyr9) were quantified 24 h following ICH. The ratios of p-Akt/Akt
and p-GSK-3β/GSK-3β were used to indicate a change in the
phosphorylation level. The phosphorylation level of Akt showed no
change following ICH, but a significant increase after rhEPO
treatment (Fig. 6A). By contrast,
the phosphorylation level of GSK-3β showed a 2.7-fold increase
(P<0.05) in response to ICH and it was reversed to approximately
the level of the sham group following rhEPO-treatment (Fig. 6B). Wortmannin pre-treatment
abolished the effect of rhEPO on the regulation of the
phosphorylation levels of Akt and GSK-3β.
Discussion
ICH remains a serious clinical problem that lacks
effective treatment. In the present study, the efficacy of rhEPO
was analyzed following hemorrhagic brain injury and the possible
signaling pathway involved in the rhEPO-mediated effect was
investigated. It was demonstrated that rhEPO treatment following
ICH resulted in a significant reduction in brain edema and
inhibition of inflammation and apoptosis that consequently led to
neurological improvement. Pharmacological intervention using
wortmannin suggested that the activation of the PI3K-Akt signaling
pathway contributed to the rhEPO-mediated beneficial effect. The
results suggested the usage of rhEPO may be a therapeutic modality
for ICH injury.
The neuroprotectivity of EPO has been demonstrated
in animal and human models under several neurological conditions,
including ischemic stroke and traumatic brain injury. This
neuroprotective effect may be due to multiple mechanisms, including
anti-apoptosis, anti-inflammation, anti-oxidation, angiogenesis and
its function as a eurotrophic factor. These effects consequently
reduce the ischemic infarct area and neuronal loss, as well as
promote cell survival following experimental insult (27,28).
In the present study, different measures were combined in an ICH
model, which further confirmed that EPO may exert its protective
effects through a variety of mechanisms at the cellular and
molecular levels and may have the potential to affect short- and
long-term clinical outcomes.
ICH initiates prominent inflammatory responses,
including the activation of resident glial cells and infiltration
of circulating inflammatory cells into the brain (3,29,30).
These inflammatory cells produce a number of proinflammatory
mediators, such as TNF-α, IL-1β and IL-6 (31). TNF-α is associated with neuronal
death and inflammation in stroke (32). IL-1β may also be involved in the
pathogenesis of ICH, since overexpression of the IL-1 receptor
antagonist significantly reduces brain edema induced by the
injection of autologous blood or thrombin into the rat striatum
(33). This suggests that the
release of proinflammatory cytokines following ICH may result in
severe secondary damage in neurons, which results in long-term
neurological deficits. Brain edema is commonly used as an indicator
of brain damage following ICH, and has been shown to be correlated
with the release of proinflammatory cytokines. Brain edema is
defined as an increase in the water content of brain tissue and is
commonly observed during the acute and subacute stages of ICH.
Studies have shown that the perihematomal edema is a result of the
local inflammatory response and contributes to poor neurological
outcomes (34,35). Moreover, anti-inflammatory
therapies attenuate brain injury (33,36).
In the present study, a rhEPO-mediated reduction of proinflammatory
cytokines was observed 1 day post-ICH, which was followed by
attenuated edema and recovery of neurological performance. It has
previously been shown that rhEPO markedly attenuates glial
activation and inhibits leukocyte infiltration in a rat model of
middle cerebral artery occlusion (37). Therefore, one possible link between
rhEPO treatment and the decreased level of proinflammatory
cytokines may be rhEPO-mediated inhibition of immune cell
activation, possibly through modulating the expression of the EPOR
on the surface of these cells. As these immune cells are important
sources of inflammatory mediators, attenuated activation results in
decreased inflammatory cytokine release and thereby inhibits the
propagation of the damaged area (38).
Although the neuroprotectivity of EPO is associated
with multiple mechanisms, including anti-inflammation and
anti-apoptosis, the molecular mechanisms underlying these effects
remain unclear. It has been shown that EPO binding to its receptor
prevents neuronal apoptosis, which involves the activation of janus
kinase 2 and the nuclear factor-κB signaling pathways (20). In addition, EPO has also been shown
to prevent apoptotic injury through an Akt-dependent mechanism
(39). Akt has been observed to
prevent cellular apoptotic degradation by inhibiting GSK-3β
activity (40,41). To the best of our knowledge, the
present study investigated for the first time the involvement of
the PI3K-Akt signaling pathway in EPO-induced neuroprotection using
the specific PI3K inhibitor wortmannin. Inhibiting the PI3K pathway
abolished the neuroprotective effect of EPO, suggesting that the
PI3K pathway is essential in EPO-induced protection against ICH.
rhEPO treatment significantly increased the protein expression of
activated Akt in the ipsilateral hemisphere 24 h following surgery,
which successively reduced the expression of activated GSK-3β.
Wortmannin treatment itself did not result in damage or exacerbate
the neurological outcome (42),
and wortmannin administration in combination with rhEPO showed no
change in the levels of the respective proteins compared with the
vehicle-treated group. These results suggest that rhEPO exerts an
anti-apoptotic effect through PI3K-Akt activation and results in
GSK-3β inhibition.
In conclusion, rhEPO treatment following ICH is
beneficial and improves neurological recovery in a rat ICH model
through a variety of cellular mechanisms, including the reduction
of edema and the inhibition of inflammation and apoptosis. In
addition, the rhEPO-mediated effect involved the activation of the
PI3K pathway.
Acknowledgements
This study was supported by grants from the Natural
Science Foundation of Chongqing (grant no. CSTC2012JJA10067). The
project was sponsored by the Municipal Educational Commission
Foundation of Chongqing (grant no. KJ110309).
References
|
1
|
Ostrowski RP, Colohan AR and Zhang JH:
Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat
model of subarachnoid hemorrhage. J Cereb Blood Flow Metab.
25:554–571. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lyden PD and Zivin JA: Hemorrhagic
transformation after cerebral ischemia: mechanisms and incidence.
Cerebrovasc Brain Metab Rev. 5:1–16. 1993.PubMed/NCBI
|
|
3
|
Zeng JS, Zheng P, Xu JF, et al: Prediction
of motor function by diffusion tensor tractography in patients with
basal ganglion haemorrhage. Arch Med Sci. 7:310–314. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Broderick J, Connolly S, Feldmann E,
Hanley D, Kase C, Krieger D, et al: Guidelines for the management
of spontaneous intracerebral hemorrhage in adults: 2007 update: a
guideline from the American Heart Association/American Stroke
Association Stroke Council, High Blood Pressure Research Council,
and the Quality of Care and Outcomes in Research Interdisciplinary
Working Group. Stroke. 38:2001–2023. 2007.
|
|
5
|
Daverat P, Castel JP, Dartigues JF and
Orgogozo JM: Death and functional outcome after spontaneous
intracerebral hemorrhage. A prospective study of 166 cases using
multivariate analysis. Stroke. 22:1–6. 1991. View Article : Google Scholar
|
|
6
|
Cotena S, Piazza O and Tufano R: The use
of erythropoietin in cerebral diseases. Panminerva Med. 50:185–192.
2008.PubMed/NCBI
|
|
7
|
Li L, Jiang Q, Ding G, et al: MRI
identification of white matter reorganization enhanced by
erythropoietin treatment in a rat model of focal ischemia. Stroke.
40:936–941. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ruscher K, Freyer D, Karsch M, et al:
Erythropoietin is a paracrine mediator of ischemic tolerance in the
brain: evidence from an in vitro model. J Neurosci. 22:10291–10301.
2002.PubMed/NCBI
|
|
9
|
Sirén AL, Fratelli M, Brines M, et al:
Erythropoietin prevents neuronal apoptosis after cerebral ischemia
and metabolic stress. Proc Natl Acad Sci USA. 98:4044–4049.
2001.PubMed/NCBI
|
|
10
|
Celik M, Gökmen N, Erbayraktar S, et al:
Erythropoietin prevents motor neuron apoptosis and neurologic
disability in experimental spinal cord ischemic injury. Proc Natl
Acad Sci USA. 99:2258–2263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hwang J, Huh J, Kim J, Jeon Y, Cho S and
Han S: Pretreatment with erythropoietin attenuates the neurological
injury after spinal cord ischaemia. Spinal Cord. 50:208–212. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiong Y, Mahmood A, Meng Y, Zhang Y, Qu C,
Schallert T and Chopp M: Delayed administration of erythropoietin
reducing hippocampal cell loss, enhancing angiogenesis and
neurogenesis, and improving functional outcome following traumatic
brain injury in rats: comparison of treatment with single and
triple dose. J Neurosurg. 113:598–608. 2010. View Article : Google Scholar
|
|
13
|
Xiong Y, Mahmood A, Lu D, et al:
Histological and functional outcomes after traumatic brain injury
in mice null for the erythropoietin receptor in the central nervous
system. Brain Res. 1230:247–257. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen G, Zhang S, Shi J, Ai J and Hang C:
Effects of recombinant human erythropoietin (rhEPO) on JAK2/STAT3
pathway and endothelial apoptosis in the rabbit basilar artery
after subarachnoid hemorrhage. Cytokine. 45:162–168. 2009.
View Article : Google Scholar
|
|
15
|
Grasso G, Buemi M, Alafaci C, et al:
Beneficial effects of sys- temic administration of recombinant
human erythropoietin in rabbits subjected to subarachnoid
hemorrhage. Proc Natl Acad Sci USA. 99:5627–5631. 2002. View Article : Google Scholar
|
|
16
|
Tseng MY, Hutchinson PJ, Richards HK, et
al: Acute systemic erythropoietin therapy to reduce delayed
ischemic deficits following aneurysmal subarachnoid hemorrhage: a
Phase II randomized, double-blind, placebo-controlled trial. J
Neurosurg. 111:171–180. 2009. View Article : Google Scholar
|
|
17
|
Chau M, Chen D and Wei L: Erythropoietin
attenuates inflammatory factors and cell death in neonatal rats
with intracerebral hemorrhage. Acta Neurochir Suppl. 111:299–305.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seyfried DM, Han Y, Yang D, Ding J and
Chopp M: Erythropoietin promotes neurological recovery after
intracerebral hemorrhage in rats. Int J Stroke. 4:250–256. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grasso G, Graziano F, Sfacteria A, et al:
Neuroprotective effect of erythropoietin and darbepoetin alfa after
experimental intracerebral hemorrhage. Neurosurgery. 65:763–770.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee ST, Chu K, Sinn DI, et al:
Erythropoietin reduces perihematomal inflammation and cell death
with eNOS and STAT3 activations in experimental intracerebral
hemorrhage. J Neurochem. 96:1728–1739. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seyfried D, Han Y, Lu D, Chen J, Bydon A
and Chopp M: Improvement in neurological outcome after
administration of atorvastatin following experimental intracerebral
hemorrhage in rats. J Neurosurg. 101:104–107. 2004. View Article : Google Scholar
|
|
22
|
Duris K, Manaenko A, Suzuki H, Rolland WB,
Krafft PR and Zhang JH: α7 nicotinic acetylcholine receptor agonist
pnu-282987 attenuates early brain injury in a perforation model of
subarachnoid hemorrhage in rats. Stroke. 42:3530–3536. 2011.
|
|
23
|
Wishka DG, Walker DP, Yates KM, Reitz SC,
Jia S, Myers JK, et al: Discovery of
N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide,
an agonist of the alpha7 nicotinic acetylcholine receptor, for the
potential treatment of cognitive deficits in schizophrenia:
synthesis and structure--activity relationship. J Med Chem.
49:4425–4436. 2006.
|
|
24
|
Aronowski J and Hall CE: New horizons for
primary intracerebral hemorrhage treatment: experience from
preclinical studies. Neurol Res. 27:268–279. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J, Li Y, Wang L, et al: Therapeutic
benefit of intravenous administration of bone marrow stromal cells
after cerebral ischemia in rats. Stroke. 32:1005–1011. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma Q, Manaenko A, Khatibi NH, Chen W,
Zhang JH and Tang J: Vascular adhesion protein-1 inhibition
provides antiinflammatory protection after an intracerebral
hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 31:881–893.
2011. View Article : Google Scholar
|
|
27
|
Del Bigio MR, Yan HJ, Buist R and Peeling
J: Experimental intracerebral hemorrhage in rats. Magnetic
resonance imaging and histopathological correlates. Stroke.
27:2312–2319. 1996.PubMed/NCBI
|
|
28
|
van der Kooij MA, Groenendaal F, Kavelaars
A, et al: Neuroprotective properties and mechanisms of
erythropoietin in in vitro and in vivo experimental models for
hypoxia/ischemia. Brain Res Rev. 59:22–33. 2008.PubMed/NCBI
|
|
29
|
Loftspring MC, McDole J, Lu A, Clark JF
and Johnson AJ: Intracerebral hemorrhage leads to infiltration of
several leukocyte populations with concomitant pathophysiological
changes. J Cereb Blood Flow Metab. 29:137–143. 2009. View Article : Google Scholar
|
|
30
|
Wang J: Preclinical and clinical research
on inflammation after intracerebral hemorrhage. Prog Neurobiol.
92:463–477. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanisch UK: Microglia as a source and
target of cytokines. Glia. 40:140–155. 2002.PubMed/NCBI
|
|
32
|
Allan SM and Rothwell NJ: Cytokines and
acute neurodegeneration. Nat Rev Neurosci. 2:734–744. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Masada T, Hua Y, Xi G, Yang GY, Hoff JT
and Keep RF: Attenuation of intracerebral hemorrhage and
thrombin-induced brain edema by overexpression of interleukin-1
receptor antagonist. J Neurosurg. 95:680–686. 2001. View Article : Google Scholar
|
|
34
|
Xi G, Keep RF and Hoff JT: Mechanisms of
brain injury after intracerebral haemorrhage. Lancet Neurol.
5:53–63. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zazulia AR, Diringer MN, Derdeyn CP and
Powers WJ: Progression of mass effect after intracerebral
hemorrhage. Stroke. 30:1167–1173. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jung KH, Chu K, Jeong SW, et al: HMG-CoA
reductase inhibitor, atorvastatin, promotes sensorimotor recovery,
suppressing acute inflammatory reaction after experimental
intracerebral hemorrhage. Stroke. 35:1744–1749. 2004. View Article : Google Scholar
|
|
37
|
Villa P, Bigini P, Mennini T, Agnello D,
et al: Erythropoietin selectively attenuates cytokine production
and inflammation in cerebral ischemia by targeting neuronal
apoptosis. J Exp Med. 198:971–975. 2003. View Article : Google Scholar
|
|
38
|
Liu X, Shen J, Jin Y, Duan M and Xu J:
Recombinant human erythropoietin (rhEPO) preconditioning on nuclear
factor-kappa B (NF-kB) activation & proinflammatory cytokines
induced by myocardial ischaemia-reperfusion. Indian J Med Res.
124:343–354. 2006.
|
|
39
|
Belayev L, Khoutorova L, Zhao W, et al:
Neuroprotective effect of darbepoetin alfa, a novel recombinant
erythropoietic protein, in focal cerebral ischemia in rats. Stroke.
36:1071–1076. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brines ML, Ghezzi P, Keenan S, et al:
Erythropoietin crosses the blood-brain barrier to protect against
experimental brain injury. Proc Natl Acad Sci USA. 97:10526–10531.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Valerio A, Bertolotti P, Delbarba A, et
al: Glycogen synthase kinase-3 inhibition reduces ischemic cerebral
damage, restores impaired mitochondrial biogenesis and prevents ROS
production. J Neurochem. 116:1148–1159. 2011. View Article : Google Scholar
|
|
42
|
Yano S, Morioka M, Fukunaga K, et al:
Activation of Akt/protein kinase B contributes to induction of
ischemic tolerance in the CA1 subfield of gerbil hippocampus. J
Cereb Blood Flow Metab. 21:351–360. 2001. View Article : Google Scholar : PubMed/NCBI
|