Introduction
Periodontal disease, a chronic inflammatory disease
of the periodontium, is initiated by bacterial infections, which
may eventually result in the loss of teeth. Porphyromonas
gingivalis (P. gingivalis), a Gram-negative and obligate
anaerobic bacterium, has long been considered to be an important
pathogen involved in human periodontal disease. Previous studies
have suggested a correlation between periodontal disease and
atherosclerosis and that P. gingivalis infection may
accelerate the development of atherosclerosis (1–4).
Lipopolysaccharide (LPS) is the main component of
the cell wall of Gram-negative bacteria (5) and has been increasingly suggested to
be responsible for the activation of vascular endothelial cells
(VECs) and the initiation of the atherosclerotic process (6,7).
P. gingivalis LPS, which is important in P.
gingivalis infection (8), may
be released into the bloodstream following scaling and root planing
or even mastication in patients with severe periodontal disease,
resulting in elevated levels of circulating endotoxin, which has
been shown to be correlated with an increased risk of
atherosclerotic incident (9–13).
It has previously been shown that P. gingivalis LPS has a
similar ability to that of Escherichia coli (E. coli)
to activate human aortic endothelial cells (HAECs) by elevating the
gene expression and protein production of vascular cell adhesion
molecule-1 (VCAM-1) (4,14). VCAM-1 belongs to the immunoglobulin
superfamily, is important in leukocyte recruitment to sites of
inflammation and accelerates the development of atherosclerosis
(15–17). Therefore, P. gingivalis
LPS-induced VCAM-1 expression may contribute to the initiation of
the atherosclerotic process.
Multiple signaling mechanisms, in a number of cell
lines, have been demonstrated to be involved in the induction of
VCAM-1 expression in response to various stimuli. This suggests
that there are divergent pathways leading to VCAM-1 expression,
depending on the nature of the stimulus and the cell type (4,18–21).
A major mechanism through which signals from extracellular stimuli
are transmitted to the nucleus involves the activation of
intracellular kinases, including those belonging to the
mitogen-activated protein kinase (MAPK) superfamily. p38 MAPK and
c-Jun N-terminal kinase (JNK) are two distinct and parallel MAPK
pathways that have been demonstrated to predominantly be involved
in the regulation of cellular inflammation in response to stimuli,
including the E. coli LPS-induced upregulation of VCAM-1
(22–26).
Previous studies have shown the activation of p38
MAPK and JNK by P. gingivalis LPS in a number of cell lines;
however, studies concerning human VECs are limited (27–29).
In addition, the majority of the previous studies investigated the
response of human umbilical VECs (HUVECs) to P. gingivalis
LPS and were, therefore, less relevant to atherosclerosis. We
recently demonstrated the involvement of the p38 MAPK pathway in
P. gingivalis LPS-induced VCAM-1 expression in HAECs
(4). However, the mechanisms
responsible for P. gingivalis LPS-induced VCAM-1 expression
in VECs have yet to be elucidated. Therefore, further study is
required to demonstrate whether the activation of JNK by P.
gingivalis LPS is associated with VCAM-1 expression in HAECs.
In addition, it is of interest that many of the genes regulated by
MAPKs are dependent on nuclear factor-κB (NF-κB) for transcription.
NF-κB has also been shown to be involved in the expression of
adhesion molecules at the transcriptional level, in various cell
types (19,20,30).
Since it has been shown that discrepancies in chemical structures,
immunobiological activities and the activation of intracellular
signaling pathways exist between P. gingivalis and E.
coli LPS (31,32), E. coli LPS was also
introduced in the present study.
The experiments in the present study were performed
to investigate the roles of JNK and NF-κB in P. gingivalis
LPS-induced VCAM-1 protein expression in HAECs. It was demonstrated
that NF-κB was required for this expression, whereas JNK was a
suppressor of VCAM-1 expression. These results provide a novel
insight into the mechanisms of P. gingivalis LPS action in
the initiation of atherosclerosis.
Materials and methods
Reagents
P. gingivalis LPS was purchased from
Invitrogen Life Technologies (San Diego, CA, USA), while E.
coli LPS (055:B5) and β-actin monoclonal antibody were obtained
from Sigma-Aldrich (St. Louis, MO, USA). The monoclonal antibodies
for JNK (56G8), phosphorylated JNK (p-JNK; G9), NF-κB, and nuclear
factor of κ light polypeptide gene enhancer in B-cells inhibitor-α
(IκB-α) were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). VCAM-1 (E-10) monoclonal antibody was obtained
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The JNK
(JNK Inhibitor II, SP600125) and NF-κB (SN50) inhibitors were
obtained from Calbiochem (San Diego, CA, USA). SP600125 was
dissolved in dimethylsulfoxide (DMSO), while SN50 was dissolved in
ultrapure water at a concentration of 50 mM for stock
solutions.
Cells and culture conditions
HAECs and Endothelial Cell Medium (ECM) were
obtained from ScienCell (Carlsbad, CA, USA). ECM was prepared for
use by mixing 5 ml Endothelial Cell Growth Supplement (ECGS), 5 ml
penicillin/streptomycin solution (P/S) and 25 ml fetal bovine serum
(FBS) into 500 ml basal medium, in accordance with the
manufacturer’s instructions. Frozen cells were thawed in a water
bath at 37°C and then seeded in 100-mm culture dishes at a density
of 5×103 cells/cm2. The cells were maintained
at 37°C in humidified 5% CO2. Twenty-four hours
subsequent to seeding, the culture medium was changed to remove the
residual DMSO and the unattached cells and was then changed every 2
days. When the cultures reached 90% confluence, the cells were
treated with 0.25% (w/v) trypsin/0.02% (w/v) EDTA until all the
cells were completely detached. The cells were subsequently
resuspended and diluted with ECM, prior to the cell suspension
being plated onto 100-mm culture dishes. The culture medium was
changed every 2 days. HAECs at 85% confluence and in the third to
sixth passages were used in all the experiments. The cells were
seeded in 60-mm culture dishes at a cell density of
5×105 cells/dish.
Preparation of cell extracts and western
blot analysis
HAECs were pretreated with the specific inhibitor
SP600125 (20 μM) or SN50 (18 mM) for 1 h prior to the addition of
LPS. Cells were harvested using centrifugation at 2,500 × g for 10
min. The collected cells were subsequently lysed with cell and
tissue protein extraction reagent containing protease inhibitor,
phosphatase inhibitor and phenylmethanesulfonyl fluoride (PMSF;
Kangchen Bio-tech, Shanghai, China). The lysates were then
centrifuged at 14,000 × g for 15 min at 4°C and the debris was
removed. The protein concentrations in the lysates were assessed
using bicinchoninic acid (BCA) reagents (Beyotime Institute of
Biotechnology, Jiangsu, China), in accordance with the
manufacturer’s protocol.
Protein samples (40 μg) were denatured and loaded
onto a 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gel, prior to being electrotransferred
onto a 0.2-μm polyvinylidene fluoride (PVDF) transfer membrane
(Pall Corp., Pensacola, FL, USA). The membranes were subsequently
blocked at room temperature with 5% (w/v) skimmed milk or 5% (w/v)
bovine serum albumin (BSA) in Tris-buffered saline and Tween 20
[TBST; Tris-HCl 50 mM, NaCl 150 mM, 0.05% (w/v) Tween-20, pH 7.4]
for ≥1 h. The blocked membranes were incubated overnight at 4°C
with the specific antibodies against VCAM-1 (1:100), β-actin
(1:5,000), p-JNK (1:2,000), JNK (1:1,000), NF-κB (1:1,000) or IκB-α
(1:1,000) in TBST. Subsequent to being washed with TBST, the
membranes were incubated with a fluorescent-labeled secondary
antibody (1:1,000) for 1 h, and the immunoreactive products were
analyzed using the Odyssey® Infrared Imaging system
(LI-COR Biosciences, Lincoln, NE, USA).
Statistical analysis
Statistical analyses were performed using an
independent samples t-test with the SPSS 20.0 statistical software
package (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
P. gingivalis LPS activated JNK signaling
pathway transduction in HAECs
To investigate whether P. gingivalis and
E. coli LPS mediated JNK phosphorylation, the activation of
this kinase was examined using an antibody specific for
tyrosine-phosphorylated JNK and western blotting. As shown in
Fig. 1A and C, the P.
gingivalis LPS-induced phosphorylation of JNK was observed
within 15 min subsequent to stimulation and reached a peak at 30
min. In E. coli LPS-treated HAECs, JNK phosphorylation was
observed as early as 5 min and reached the highest level at 60 min,
which was the end of the observation period (Fig. 1B and C).
Involvement of JNK in P. gingivalis
LPS-induced VCAM-1 expression
To demonstrate the involvement of the JNK cell
signaling pathway in P. gingivalis and E. coli
LPS-induced VCAM-1 expression in HAECs, SP600125, a selective
pharmacological inhibitor (33),
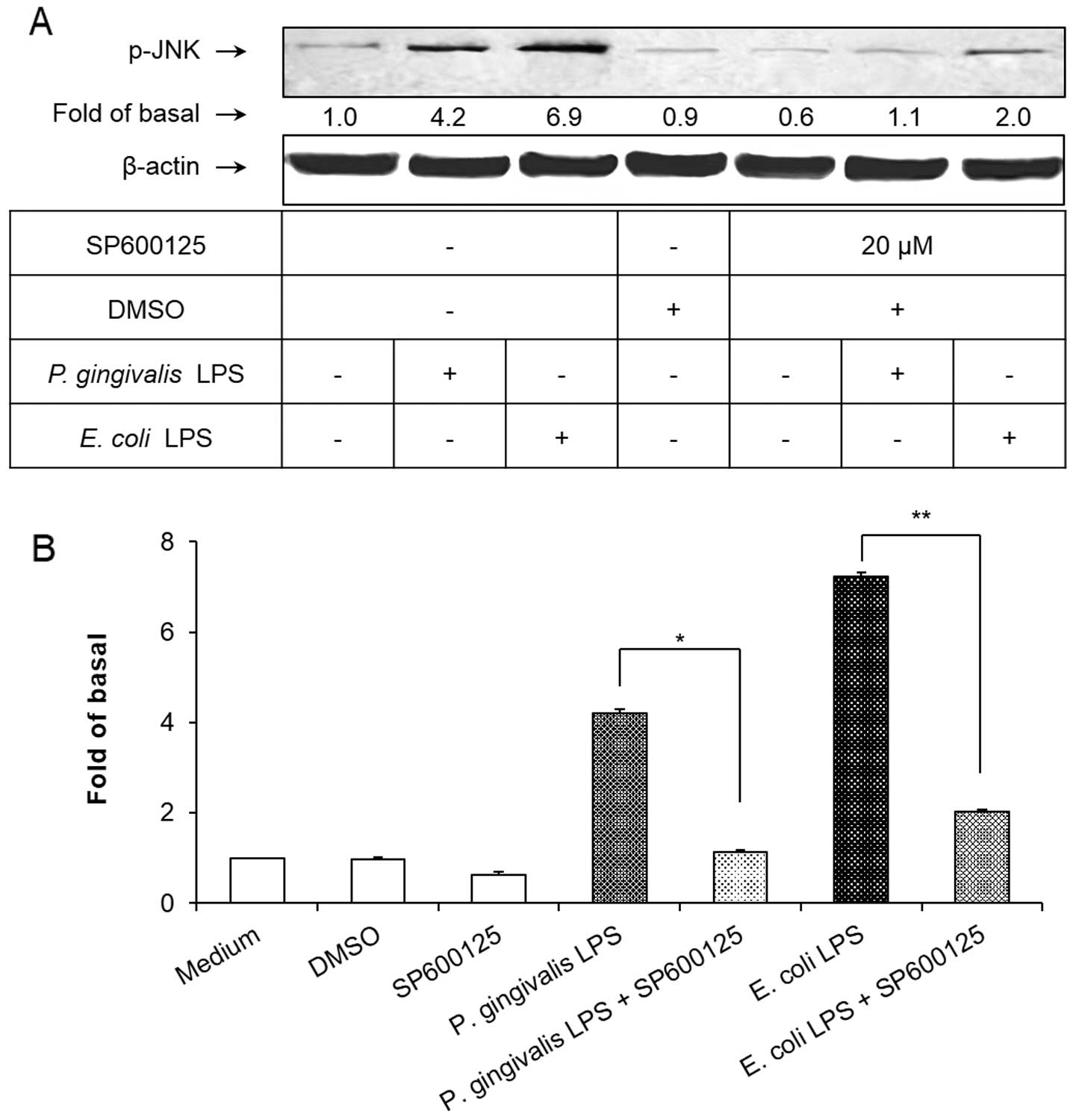
was used to block the JNK signaling pathway. As shown in Fig. 2, pretreatment of the HAECs with
SP600125 at a concentration of 20 μM for 1 h significantly
inhibited the P. gingivalis and E. coli LPS-induced
JNK phosphorylation (P<0.01). However, SP600125 failed to
suppress the upregulation of VCAM-1 induced by the P.
gingivalis and E. coli LPS. Notably, blocking the
intracellular JNK signaling pathway with SP600125 significantly
elevated the expression of VCAM-1 in LPS-treated HAECs (P<0.01;
Fig. 3).
P. gingivalis LPS-activated NF-κB
signaling pathway transduction in HAECs
To determine the activation of the transcription
factor NF-κB in HAECs exposed to P. gingivalis and E.
coli LPS, western blotting was used to assess the degradation
of IκB-α, an indicator of NF-κB activation, as well as the
activation pattern of p65, a component of NF-κB, using antibodies
specific for IκB-α and NF-κB p65, respectively. As shown in
Fig. 4, IκB-α was significantly
degraded, corresponding with the upregulation of NF-κB p65,
following 30 min of exposure of the HAECs to P. gingivalis
and E. coli LPS. SN50, a synthetic cell permeable peptide
and inhibitor of NF-κB (34), has
been shown to significantly attenuate the degradation of IκB-α and
the elevated expression of NF-κB p65.
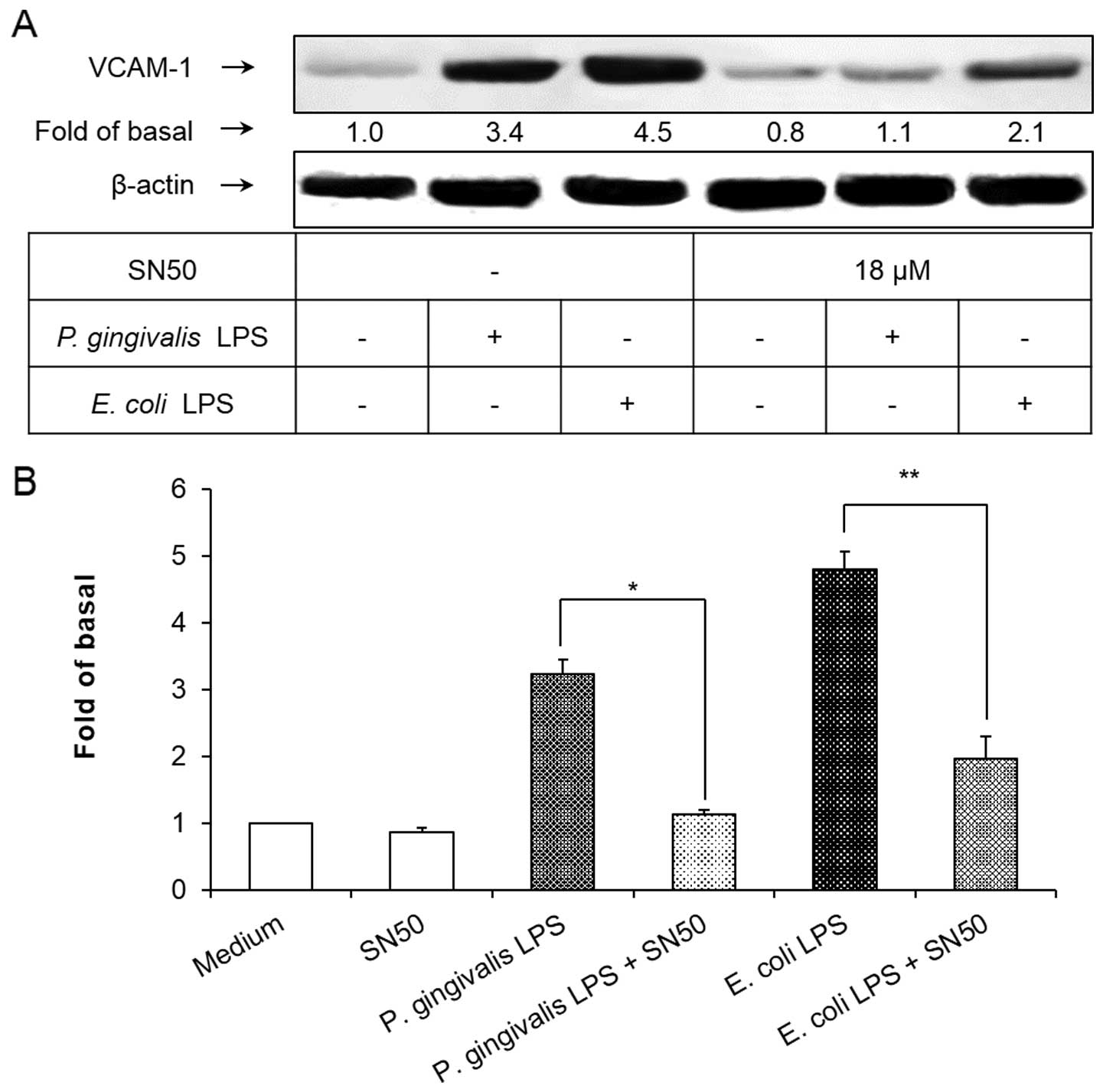
NF-κB inhibitor suppresses P. gingivalis
LPS-induced VCAM-1 expression
To demonstrate the involvement of the NF-κB
signaling pathway in P. gingivalis and E. coli
LPS-induced VCAM-1 expression in HAECs, SN50 was used to block the
NF-κB signaling pathway. As shown in Fig. 5, pretreatment of the HAECs with
SN50 at a concentration of 18 μM for 1 h significantly inhibited
the induction of VCAM-1 expression in HAECs exposed to P.
gingivalis and E. coli LPS. These results suggested that
the activation of NF-κB was essential for the P. gingivalis
and E. coli LPS-induced VCAM-1 expression in HAECs.
Discussion
Mononuclear cell (MNC) adhesion to the
atherosclerosis-prone vascular endothelium has been suggested to be
the initial step of atherosclerosis (15–17).
The upregulation of VCAM-1 on the surface of the VECs may be
important in the recruitment, rolling, firm adhesion and
infiltration of MNCs at sites of inflammation in the vascular
endothelium and may accelerate the development of atherosclerosis
(35). P. gingivalis LPS
has been shown to be an inducer of VCAM-1 (4,14);
however, little is known about the intracellular signaling pathways
leading to VCAM-1 expression in VECs exposed to P.
gingivalis LPS. In this study, we investigated the involvement
of the JNK and NF-κB signaling pathways in P. gingivalis
LPS-induced VCAM-1 expression in HAECs. Using western blotting, we
demonstrated that pretreatment of the HAECs with a JNK inhibitor,
SP600125, significantly attenuated JNK phosphorylation; however, an
upregulation, instead of a downregulation, of P. gingivalis
LPS-induced VCAM-1 expression was observed in SP600125 pretreated
HAECs. Furthermore, the activation of NF-κB by P. gingivalis
LPS in the HAECs resulted in VCAM-1 expression, since the
activation of NF-κB and subsequent expression of VCAM-1 was
inhibited by an inhibitory peptide of NF-κB, SN50. These results
suggest that NF-κB was essential for the induction of VCAM-1
protein production in the HAECs exposed to P. gingivalis
LPS, whereas JNK may be a suppressor of VCAM-1 expression in
HAECs.
There are at least three distinct and parallel MAPK
pathways, including p38 MAPK, JNK and the extracellular
signal-regulated kinase (ERK) pathway (25). It has been demonstrated that the
p38 MAPK and JNK pathways mediate signaling stimulated by bacterial
components (LPS, lipoteichoic acid), inflammatory cytokines
[interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α)] and
stress factors (osmotic shock, heat shock,
H2O2, UV radiation and DNA-damaging agents),
while activation of ERK is important in mediating cell
proliferation in response to growth factors and mitogens (36–38).
We recently demonstrated that p38 MAPK mediated P.
gingivalis LPS-induced VCAM-1 expression in HAECs (4). Therefore, the involvement of JNK in
this effect was investigated in the present study.
In this study, we demonstrated the activation of the
JNK pathway in P. gingivalis and E. coli
LPS-stimulated HAECs. As shown in Fig.
1, P. gingivalis LPS-induced JNK phosphorylation was
weaker and reached a peak much earlier than that induced by E.
coli LPS. Similar results were observed in LPS-induced VCAM-1
expression and NF-κB activation (Figs.
3–5). Previous studies have
revealed discrepancies in the chemical structure of lipid A, the
biologically active center of LPS, between P. gingivalis and
E. coli LPS (32). In
addition, it has previously been demonstrated that the endotoxic
activities of P. gingivalis LPS and its lipid A are
relatively weak when compared with those of E. coli-derived
LPS and lipid A (4,13,32).
The observations in the present study were consistent with the
results from those studies, suggesting the potential involvement of
P. gingivalis LPS in chronic inflammatory processes, such as
atherosclerosis and periodontal disease, rather than acute
infection, such as sepsis.
Although the present and previous studies have
demonstrated the activation of the JNK pathway by P.
gingivalis and E. coli LPS (26,39),
the correlation between the activation of JNK and the expression of
VCAM-1 has been contradictory in different cell lines. It has been
shown that the JNK pathway mediated VCAM-1 expression in human
tracheal smooth muscle cells (HTSMCs) exposed to TNF-α (19) and in endothelial cells (EVC304)
exposed to E. coli LPS (21). However, Binion et
al(20) demonstrated that in
human intestinal microvascular endothelial cells (HIMECs)
activation of JNK was not a requisite for the induction of VCAM-1
production by E. coli LPS. These results suggest that VCAM-1
is selectively expressed in a cell type and stimulus-specific
manner and is mediated through the activation of diverse
intracellular signaling pathways. Furthermore, the cell lines used
in the majority of the previous studies concerning the activation
of JNK in endothelial cells exposed to LPS were venous VECs
(20,21). It has been demonstrated that there
are genetic variances between artery and vein-derived endothelial
cells, which contribute to the different biological and
immunological responses to atherosclerotic factors (13,40,41).
Of note was the fact that the HUVEC cell line used in a number of
studies was the EVC304 cell line (21,42),
which has been revealed to be genetically identical to the human
bladder cancer-derived epithelial cell line T24/83 and has been
suggested to be inappropriate for the study of endothelial cell
biology (43). The involvement of
the JNK pathway in LPS-induced VCAM-1 expression in HAECs remains
to be determined. As shown in Fig.
2, we elucidated that SP600125, an anthrapyrazole and a
reversible ATP-competitive inhibitor of JNK, significantly
attenuated the phosphorylation of JNK. However, this was
demonstrated to enhance the P. gingivalis and E. coli
LPS-induced protein production of VCAM-1 in HAECs (Fig. 3). These observations suggested that
JNK may be an intracellular suppressor for the expression of VCAM-1
in HAECs.
The observation that SP600125 enhanced VCAM-1
expression indicated that the administration of SP600125 with P.
gingivalis and E. coli LPS caused an additive effect on
VCAM-1 expression in the HAECs (Fig.
3). This result was consistent with those of previous studies,
in which SP600125 was shown to elicit additive effects with TNF-α
on VCAM-1 expression in human chondrosarcoma cells and in gingival
fibroblasts (44,45), although a study using HK-2 cells
revealed contradictory observations (46). There have been few studies
concerning the additive effect of SP600125; therefore, further
investigation is required to fully elucidate the underlying
molecular mechanism of JNK in VCAM-1 expression.
As extensively studied previously, inflammatory
responses following exposure to extracellular stimuli highly depend
on the activation of the transcription factor NF-κB (47–50).
The sequestration of NF-κB by IκB-α in the cytoplasm and the
phosphorylation of IκB-α, leading to the proteasomal degradation of
IκB-α, results in the activation and translocation of NF-κB into
the nucleus, a process that is essential in the expression of a
number of genes, such as adhesion molecules, in various cell types
(14,48,50).
In the present study, as shown in Fig.
4, the degradation of IκB-α and the activation of NF-κB p65
were observed following P. gingivalis LPS exposure and were
also observed to be inhibited by SN50. Furthermore, P.
gingivalis LPS-induced VCAM-1 expression was almost completely
suppressed by SN50 (Fig. 5),
indicating that the activation of NF-κB was essential for the
induction of VCAM-1 expression in P. gingivalis-stimulated
HAECs.
Within the limitations of this study, we have
demonstrated that P. gingivalis LPS has the ability to
accelerate the development of atherosclerosis by upregulating the
expression of VCAM-1 in HAECs, a process that is critical in the
initiation of atherosclerosis. Furthermore, we demonstrated that
P. gingivalis LPS was able to activate the JNK and NF-κB
signaling pathways in HAECs. The activation of the NF-κB pathway
was essential for the induction of VCAM-1 expression in HAECs
exposed to P. gingivalis LPS, whereas JNK may be a
suppressor of VCAM-1 expression. To the best of our knowledge, this
study is the first to investigate the role of JNK in P.
gingivalis LPS-induced VCAM-1 expression in HAECs.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 81100753) and the Science
and Technology Commission of Shanghai Municipality (no.
09JC1409100).
References
|
1
|
Lalla E, Lamster IB, Hofmann MA, et al:
Oral infection with a periodontal pathogen accelerates early
atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb
Vasc Biol. 23:1405–1411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cavrini F, Sambri V, Moter A, et al:
Molecular detection of Treponema denticola and
Porphyromonas gingivalis in carotid and aortic atheromatous
plaques by FISH: report of two cases. J Med Microbiol. 54:93–96.
2005.
|
|
3
|
Zhang MZ, Li CL, Jiang YT, et al:
Porphyromonas gingivalis infection accelerates intimal
thickening in iliac arteries in a balloon-injured rabbit model. J
Periodontol. 79:1192–1199. 2008. View Article : Google Scholar
|
|
4
|
Liu B, Cheng L, Liu D, et al: Role of p38
mitogen-activated protein kinase pathway in Porphyromonas
gingivalis lipopolysaccharide-induced VCAM-1 expression in
human aortic endothelial cells. J Periodontol. 83:955–962. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raetz CR: Biochemistry of endotoxins. Annu
Rev Biochem. 59:129–170. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rice JB, Stoll LL, Li WG, et al: Low-level
endotoxin induces potent inflammatory activation of human blood
vessels: inhibition by statins. Arterioscler Thromb Vasc Biol.
23:1576–1582. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stoll LL, Denning GM and Weintraub NL:
Potential role of endotoxin as a proinflammatory mediator of
atherosclerosis. Arterioscler Thromb Vasc Biol. 24:2227–2236. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Holt SC, Kesavalu L, Walker S and Genco
CA: Virulence factors of Porphyromonas gingivalis.
Periodontol 2000. 20:168–238. 1999. View Article : Google Scholar
|
|
9
|
Geerts SO, Nys M, De MP, et al: Systemic
release of endotoxins induced by gentle mastication: association
with periodontitis severity. J Periodontol. 73:73–78. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ide M, Jagdev D, Coward PY, Crook M,
Barclay GR and Wilson RF: The short-term effects of treatment of
chronic periodontitis on circulating levels of endotoxin,
C-reactive protein, tumor necrosis factor-alpha, and interleukin-6.
J Periodontol. 75:420–428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pussinen PJ, Vilkuna-Rautiainen T, Alfthan
G, et al: Severe periodontitis enhances macrophage activation via
increased serum lipopolysaccharide. Arterioscler Thromb Vasc Biol.
24:2174–2180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lafaurie GI, Mayorga-Fayad I, Torres MF,
et al: Periodontopathic microorganisms in peripheric blood after
scaling and root planing. J Clin Periodontol. 34:873–879. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Erridge C, Spickett CM and Webb DJ:
Non-enterobacterial endotoxins stimulate human coronary artery but
not venous endothelial cell activation via Toll-like receptor 2.
Cardiovasc Res. 73:181–189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakamura N, Yoshida M, Umeda M, et al:
Extended exposure of lipopolysaccharide fraction from
Porphyromonas gingivalis facilitates mononuclear cell
adhesion to vascular endothelium via Toll-like receptor-2 dependent
mechanism. Atherosclerosis. 196:59–67. 2008.PubMed/NCBI
|
|
15
|
Fries JW, Williams AJ, Atkins RC, Newman
W, Lipscomb MF and Collins T: Expression of VCAM-1 and E-selectin
in an in vivo model of endothelial activation. Am J Pathol.
143:725–737. 1993.PubMed/NCBI
|
|
16
|
Cybulsky MI, Iiyama K, Li H, et al: A
major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J
Clin Invest. 107:1255–1262. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ley K and Huo Y: VCAM-1 is critical in
atherosclerosis. J Clin Invest. 107:1209–1210. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang CC, Lin WN, Lee CW, et al:
Involvement of p42/p44 MAPK, p38 MAPK, JNK, and NF-kappaB in
IL-1beta-induced VCAM-1 expression in human tracheal smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol. 288:L227–L237. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee CW, Lin WN, Lin CC, et al:
Transcriptional regulation of VCAM-1 expression by tumor necrosis
factor-alpha in human tracheal smooth muscle cells: involvement of
MAPKs, NF-kappaB, p300, and histone acetylation. J Cell Physiol.
207:174–186. 2006. View Article : Google Scholar
|
|
20
|
Binion DG, Heidemann J, Li MS, Nelson VM,
Otterson MF and Rafiee P: Vascular cell adhesion molecule-1
expression in human intestinal microvascular endothelial cells is
regulated by PI 3-kinase/Akt/MAPK/NF-kappaB: inhibitory role of
curcumin. Am J Physiol Gastrointest Liver Physiol. 297:G259–G268.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shan Y, Lin N, Yang X, et al:
Sulphoraphane inhibited the expressions of intercellular adhesion
molecule-1 and vascular cell adhesion molecule-1 through
MyD88-dependent toll-like receptor-4 pathway in cultured
endothelial cells. Nutr Metab Cardiovasc Dis. 22:215–222. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han J, Lee JD, Bibbs L and Ulevitch RJ: A
MAP kinase targeted by endotoxin and hyperosmolarity in mammalian
cells. Science. 265:808–811. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang Y, Chen C, Li Z, et al:
Characterization of the structure and function of a new
mitogen-activated protein kinase (p38beta). J Biol Chem.
271:17920–17926. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang Y, Gram H, Zhao M, et al:
Characterization of the structure and function of the fourth member
of p38 group mitogen-activated protein kinases, p38delta. J Biol
Chem. 272:30122–30128. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schaeffer HJ and Weber MJ:
Mitogen-activated protein kinases: specific messages from
ubiquitous messengers. Mol Cell Biol. 19:2435–2444. 1999.PubMed/NCBI
|
|
26
|
Lin WN, Luo SF, Lee CW, Wang CC, Wang JS
and Yang CM: Involvement of MAPKs and NF-kappaB in LPS-induced
VCAM-1 expression in human tracheal smooth muscle cells. Cell
Signal. 19:1258–1267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Watanabe K, Yilmaz O, Nakhjiri SF, Belton
CM and Lamont RJ: Association of mitogen-activated protein kinase
pathways with gingival epithelial cell responses to
Porphyromonas gingivalis infection. Infect Immun.
69:6731–6737. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choi EK, Park SA, Oh WM, et al: Mechanisms
of Porphyromonas gingivalis-induced monocyte chemoattractant
protein-1 expression in endothelial cells. FEMS Immunol Med
Microbiol. 44:51–58. 2005.
|
|
29
|
Lee SD, Wu CC, Kuo WW, et al:
Porphyromonas gingivalis-related cardiac cell apoptosis was
majorly co-activated by p38 and extracellular signal-regulated
kinase pathways. J Periodontal Res. 41:39–46. 2006. View Article : Google Scholar
|
|
30
|
Lazaar AL, Albelda SM, Pilewski JM,
Brennan B, Pure E and Panettieri RA Jr: T lymphocytes adhere to
airway smooth muscle cells via integrins and CD44 and induce smooth
muscle cell DNA synthesis. J Exp Med. 180:807–816. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hamada S, Koga T, Nishihara T, Fujiwara T
and Okahashi N: Characterization and immunobiologic activities of
lipopolysaccharides from periodontal bacteria. Adv Dent Res.
2:284–291. 1988.PubMed/NCBI
|
|
32
|
Ogawa T and Yagi T: Bioactive mechanism of
Porphyromonas gingivalis lipid A. Periodontol 2000.
54:71–77. 2010.
|
|
33
|
Bennett BL, Sasaki DT, Murray BW, et al:
SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase.
Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin YZ, Yao SY, Veach RA, Torgerson TR and
Hawiger J: Inhibition of nuclear translocation of transcription
factor NF-kappa B by a synthetic peptide containing a cell
membrane-permeable motif and nuclear localization sequence. J Biol
Chem. 270:14255–14258. 1995. View Article : Google Scholar
|
|
35
|
Muller WA: Mechanisms of leukocyte
transendothelial migration. Annu Rev Pathol. 6:323–344. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eder J: Tumour necrosis factor alpha and
interleukin 1 signalling: do MAPKK kinases connect it all? Trends
Pharmacol Sci. 18:319–322. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Robinson MJ and Cobb MH: Mitogen-activated
protein kinase pathways. Curr Opin Cell Biol. 9:180–186. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang CM, Luo SF, Wang CC, et al: Tumour
necrosis factor-alpha- and interleukin-1beta-stimulated cell
proliferation through activation of mitogen-activated protein
kinase in canine tracheal smooth muscle cells. Br J Pharmacol.
130:891–899. 2000. View Article : Google Scholar
|
|
39
|
Diya Z, Lili C, Shenglai L, Zhiyuan G and
Jie Y: Lipopolysaccharide (LPS) of Porphyromonas gingivalis
induces IL-1beta, TNF-alpha and IL-6 production by THP-1 cells in a
way different from that of Escherichia coli LPS. Innate
Immun. 14:99–107. 2008.
|
|
40
|
Swift MR and Weinstein BM: Arterial-venous
specification during development. Circ Res. 104:576–588. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu X, Zou Y, Liang Y, et al: COUP-TFII
switches responses of venous endothelium to atherosclerotic factors
through controlling the profile of various inherent genes
expression. J Cell Biochem. 112:256–264. 2011. View Article : Google Scholar
|
|
42
|
Zhang D, Zheng H, Zhao J, et al:
Porphorymonas gingivalis induces intracellular adhesion
molecule-1 expression in endothelial cells through the nuclear
factor-kappaB pathway, but not through the p38 MAPK pathway. J
Periodontal Res. 46:31–38. 2011. View Article : Google Scholar
|
|
43
|
Brown J, Reading SJ, Jones S, et al:
Critical evaluation of ECV304 as a human endothelial cell model
defined by genetic analysis and functional responses: a comparison
with the human bladder cancer derived epithelial cell line T24/83.
Lab Invest. 80:37–45. 2000. View Article : Google Scholar
|
|
44
|
Ju JW, Kim SJ, Jun CD and Chun JS: p38
kinase and c-Jun N-terminal kinase oppositely regulates tumor
necrosis factor alpha-induced vascular cell adhesion molecule-1
expression and cell adhesion in chondrosarcoma cells. IUBMB Life.
54:293–299. 2002. View Article : Google Scholar
|
|
45
|
Hosokawa Y, Hosokawa I, Ozaki K, Nakae H
and Matsuo T: Cytokines differentially regulate ICAM-1 and VCAM-1
expression on human gingival fibroblasts. Clin Exp Immunol.
144:494–502. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ho AW, Wong CK and Lam CW: Tumor necrosis
factor-alpha up-regulates the expression of CCL2 and adhesion
molecules of human proximal tubular epithelial cells through MAPK
signaling pathways. Immunobiology. 213:533–544. 2008. View Article : Google Scholar
|
|
47
|
Newton R, Kuitert LM, Bergmann M, Adcock
IM and Barnes PJ: Evidence for involvement of NF-kappaB in the
transcriptional control of COX-2 gene expression by IL-1beta.
Biochem Biophys Res Commun. 237:28–32. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen CC, Chen JJ and Chou CY: Protein
kinase calpha but not p44/42 mitogen-activated protein kinase, p38,
or c-Jun NH(2)-terminal kinase is required for intercellular
adhesion molecule-1 expression mediated by interleukin-1beta:
involvement of sequential activation of tyrosine kinase, nuclear
factor-kappaB-inducing kinase, and IkappaB kinase 2. Mol Pharmacol.
58:1479–1489. 2000.
|
|
49
|
Guha M and Mackman N: LPS induction of
gene expression in human monocytes. Cell Signal. 13:85–94. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bian ZM, Elner SG, Yoshida A, Kunkel SL,
Su J and Elner VM: Activation of p38, ERK1/2 and NIK pathways is
required for IL-1beta and TNF-alpha-induced chemokine expression in
human retinal pigment epithelial cells. Exp Eye Res. 73:111–121.
2001. View Article : Google Scholar : PubMed/NCBI
|