Introduction
Traumatic brain injury (TBI) is a major source of
mortality and disability worldwide (1). Usually, the initial mechanical insult
(primary injury) is limited and focal. However, these injuries
progressively enlarge in extent and severity by mechanisms of
secondary injury, the most significant of which are hypoxia,
ischemia and edema, which occur within minutes or weeks following
the primary injury (1–4). Compared with the primary injury,
secondary injuries to the brain are more progressive and may
ultimately be the deciding factors in patient recovery (1).
Pathophysiological procedures, including hypoxia,
ischemia and edema, are interrelated and mutually improved. The
brain consumes almost 20% of the total oxygen utilized by the body
at rest. Due to its high metabolic rate, the brain requires more
oxygen and glucose than other organs and is extremely vulnerable to
ischemia or hypoxia resulting from decreased cerebral blood flow
(CBF), increased intracranial pressure (ICP) and/or swelling and
edema (2). Cerebral edema is a
fairly common pathophysiological entity among TBI patients and is
defined as an excess accumulation of water in the intra- and/or
extra-cellular spaces of the brain, which can be induced by
vasogenic, cytotoxic or osmotic factors (1,5–7). TBI
may result in interstitial (vasogenic) and intracellular
(cytotoxic) edema, which contribute to secondary injury (1–3,7).
Aside from the forces incurred by the initial trauma, the prolonged
compressive forces of cerebral edema may further impair brain
function by distorting brain tissues, elevating ICP and reducing
CBF, which aggravates ischemia and hypoxia of the brain (1,2,7).
Due to its selective vulnerability, the hippocampus
is particularly sensitive to ischemia or edema of secondary injury
following TBI (8–12). Damage to the hippocampal region
results in neuronal apoptosis and leads to cognitive, memory and
learning impairments (8,10,13,14).
Therefore, it is crucial to protect hippocampal cells from the
insult of ischemia, hypoxia and edema, in order to improve the
outcome of TBI. Aquaporins (AQPs) are water channels that provide
the major route for water movement across plasma membranes in a
variety of tissues, including the brain (6,15–20).
As a member of the AQPs, AQP-1 has been found to be expressed in
astrocytes in cerebral edema associated with TBI, indicating an
intimate connection between AQP-1 and cerebral edema (21). In the present study, a series of
assays to determine APQ-1 expression in the hippocampal region of
mouse models were designed and conducted, and the role of AQP-1 in
the hippocampal damage of secondary injury following TBI was
evaluated.
Materials and methods
Animal preparation
All experimental animals were purchased from the
Department of Experimental Animal of China Medical University
(Shenyang, China). The animals were handled according to the
guidelines of the Council for International Organization of Medical
Sciences on Animal Experimentation (World Health Organization,
Geneva, Switzerland) and the China Medical University. Animal
protocols were approved by the Institutional Animal Care and Use
Committee (China Medical University). A total of 150 male BALB/c
mice (8–10 weeks-old), with an average body weight of 20–25 g, were
housed in cages in a light/dark cycle of 12 h. The animals were
randomly divided into a control group (sham, n=75) and an
experimental group (TBI, n=75); each group contained three
subgroups according to sacrifice time (6, 24 and 72 h, n=25)
following closed craniocerebral injury (CCI).
Establishment of animal models
Experimental CCI was induced using a modified
weight-drop device, as described by Chen et al (22,23).
Briefly, following induction of 4–5% isoflurane anesthesia, a
midline longitudinal incision was performed, the skin was retracted
and the skull was exposed. The left anterior frontal area was
identified as the impact area and a Teflon tipped cone (diameter, 2
mm) was placed 1.5 mm lateral to the midline in the mid-coronal
plane. The head was fixed and a 40-g weight was dropped onto the
cone from a height of 25 cm, resulting in a focal injury to the
left hemisphere. Following trauma, the mice received supporting
oxygenation with 95% O2 for no longer than 2 min and
were then returned to their cages. The scalp wound was closed using
standard suture material and the wound area was treated with
lidocaine cream. Sham controls received anesthesia and skin
incision only.
Neurological function score of mouse
models
The neurological function of each mouse model was
evaluated at various times (6, 24 and 72 h) following CCI using a
modified neurological severity score (NSS) as previously described
(22,23). Table
I shows a set of modified NSS. Neurological function was graded
on a scale of 0 to 18 (normal score, 0; maximal deficit score, 18).
NSS is a composite of motor, sensory, reflex and balance tests. In
the severity scores of injury, 1 score point is awarded for the
inability to perform the test or for the lack of a tested reflex;
thus, the higher the score, the more severe the injury. Thus, a
score of 13–18 indicates a ‘severe’ injury, while a score of 7–12
reflects a ‘moderate’ injury and <6 indicates a ‘mild’ injury.
Initial severity of the trauma was assessed 1 h following trauma.
The parameters of damage for severe injury were established in the
present study. A total of 20 mice from each subgroup in the
experimental or control groups were randomly selected for each
assay (four for edema evaluation, eight for immunochemistry and
apoptosis assays and eight for western blot analysis).
 | Table IModified neurological severity score
for mice. |
Table I
Modified neurological severity score
for mice.
| Characteristic | Point |
|---|
| Motor tests | (6) |
| Raising rat by the
tail | (3) |
| Flexion of
forelimb | 1 |
| Flexion of
hindlimb | 1 |
| Head moved
>10° to vertical axis within 30 sec | 1 |
| Placing rat on the
floor | (3) |
| Normal walk | 0 |
| Inability to walk
straight | 1 |
| Circling toward
the paretic side | 2 |
| Fall down to the
paretic side | 3 |
| Sensory tests | (2) |
| Placing test
(visual and tactile test) | 1 |
| Proprioceptive
test (deep sensation, pushing the paw against the table edge to
stimulate limb muscles) | 2 |
| Beam balance
tests | (6) |
| Balances with
steady posture | 0 |
| Grasps side of
beam | 1 |
| Hugs the beam and
one limb falls down from the beam | 2 |
| Hugs the beam and
two limbs fall down from the beam or spins on beam (>60
sec) | 3 |
| Attempts to
balance on the beam but falls off (>40 sec) | 4 |
| Attempts to
balance on the beam but falls off (<20 sec) | 5 |
| Falls off: No
attempt to balance or hang on to the beam (<20 sec) | 6 |
| Reflexes absent and
abnormal movements | (4) |
| Pinna reflex (head
shake when touching the auditory meatus) | 1 |
| Corneal reflex
(eye blink when lightly touching the cornea with cotton) | 1 |
| Startle reflex
(motor response to a brief noise from snapping a clipboard
paper) | 1 |
| Seizures,
myoclonus, myodystony | 1 |
Evaluation of edema
As previously described, cerebral edema was measured
by detecting the tissue water content in the ipsilateral
hippocampus (22,24). Following anesthesia using
intraperitoneal administration of 0.8% pentobarbital (40 mg/kg), a
mouse was sacrificed by decapitation and the left hippocampus was
rapidly removed. A tissue segment (~20 mg) was taken from the left
hippocampus and weighed to yield wet weight (WW). Following
desiccation oven treatment for 24 h at 95°C, the sample was
reweighed to yield dry weight (DW). The calculation formula was as
follows: Tissue water content (%) = [(WW − DW) × 100]/WW. Edema was
measured at 6, 24 and 72 h following CCI in four mice at each time
point.
Tissue preparation and
immunohistochemistry assay
At 6, 24 and 72 h following sham surgery or CCI,
mice in the sham or TBI groups were anesthetized, sacrificed by
decapitation and the whole left hippocampus was rapidly removed and
separated on ice. The left hippocampus of each mouse was subject to
immunochemistry, apoptosis and western blot analysis.
For immunochemistry and apoptosis detection, the
left hippocampus from each mouse was placed in 4% paraformaldehyde
in phosphate-buffered saline [PBS; 0.1 M, (pH 7.6)], containing
0.1% diethylpyrocarbonate. The tissue was stored in the same
fixative solution for 4–6 h at 4°C and then placed in a 30%
phosphate-buffered sucrose at 4°C until immersion. The tissue was
then paraffin-embedded and 8-μm thick coronal sections were created
using a rotary microtome (Leica RM2235; Leica Microsystems GmbH,
Wetzlar, Germany). Sections were stored at −80°C for later use.
A third series of every tenth section of each left
hemisphere was used for detection and quantification of AQP-1
staining. Immunohistochemistry was performed according to the
manufacturer’s instructions of the Histostain-SP kit (Invitrogen
Life Technologies, Carlsbad, CA, USA) and the primary antibody
(rabbit anti-mouse AQP-1; Sigma-Aldrich, St. Louis, MO, USA) was
diluted at 1:200. AQP-1 binding was visualized using the standard
avidin/biotinylated enzyme complex-horseradish peroxidase (HRP)
staining procedure, with 3,3′-diaminobenzidine (DAB) as a
chromogen. In the control sections, the primary antibody was
replaced with normal goat serum. The sections were inspected using
an optic microscope (BX40; Olympus, Tokyo, Japan). All experimental
groups were included in each immunochemical analysis and were,
therefore, processed under the same conditions.
Terminal deoxynucleotidyl transferase
(TdT)-mediated dUTP nick-end labeling (TUNEL) staining of
hippocampal tissue
Apoptosis of hippocampal cells was detected using an
in situ cell death detection kit (Roche Diagnostics,
Mannheim, Germany) according to the manufacturer’s instructions.
Briefly, 8-μm paraffin sections underwent conventional
deparaffinization and hydration and were then were immersed in 3%
H2O2 to quench endogenous peroxidase
activity, followed by incubation at room temperature for 15 min
with proteinase K [20 μg/ml in Tris/HCl (pH 7.4–8.0); Promega
Corporation, Madison, WI, USA]. Thereafter, sections were incubated
with 50 μl TUNEL reaction mixture (TdT and labeled nucleotide
mixture) for 1 h in 37°C. The sections were rinsed in PBS, wiped
dry, incubated with 50 μl converter-peroxidase for 30 min at 37°C,
washed again and further incubated with 50 μl DAB substrate
solution (Roche Diagnostics) at room temperature for 10 min. The
sections were subsequently dehydrated, cleared and mounted. Cells
containing fragmented nuclear chromatin exhibited a brown nuclear
stain. As negative controls, sections were processed without TdT
buffer. Apoptotic cells were examined with an optic microscope
(BX40).
Western blot analysis
For western blot analysis, the left hippocampus
removed from each mouse was used for sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western
blot analysis, as described previously (25). Equal quantities of tissue lysates
were resolved by SDS-PAGE, transferred onto polyvinylidene fluoride
membranes (Roche Diagnostics) using electroblotting, probed with
specific primary antibodies followed by secondary antibody
conjugation and then analyzed. The primary antibodies were
monoclonal rabbit anti-mouse AQP-1 (1:500; Sigma-Aldrich) and
monoclonal anti-β-actin antibody (1:500; Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA) and were detected using HRP-conjugated
goat anti-mouse IgG (1:500; Sigma-Aldrich) in blocking solution.
The non-glycosylated (28 kDa) band was used for analysis of changes
in AQP-1 expression in all experiments. Immunoreactive protein
bands were visualized with enhanced chemiluminescence reagent
(ECL-PLUS; Amersham Biosciences, Piscataway, NJ, USA), scanned with
a FujiFilm LAS300 Intelligent Dark Box scanner (Fujifilm, Tokyo,
Japan) and quantified for pixel density by the optical density (OD)
method using Image J software (version 1.46; http://rsb.info.nih.gov/ij/).
Statistical analysis
All analyses were performed in triplicate and the
representative results are presented as the mean ± standard
deviation (SD). The data were analyzed using SPSS for Windows
software (version 18.0; SPSS, Inc., Chicago, IL, USA). Comparisons
between groups were made with a one-way analysis of variance and a
paired t-test. P<0.05 or P<0.01 were considered to indicate a
statistically significant difference.
Results
Outcomes of mouse models
In a preliminary experiment, the weight and distance
selected for the object in the weight-drop device were repeatedly
tested to guarantee that the average NSS score for the majority of
mouse models was confined to the range of severe injury (score,
13–18). The clinical statuses of the injured mice were evaluated by
the modified NSS at 6, 24 and 72 h following CCI. The average NSS
was found to decrease from 15.3±0.9 at 1 h, to 14.2±1.1 at 6 h,
11.2±0.6 at 24 h and 10.6±0.8 at 72 h following CCI. In the control
group, mice that received sham surgery were able to perform all
tasks and obtained a score of 0 in the NSS.
Edema formation
The experimental CCI resembled cerebral edema
following TBI. In the control group, the average cerebral water
content in the left hippocampi at various times showed no marked
fluctuation (77.23±0.46%, 77.51±0.35% and 77.48±0.51% at 6, 24 and
72 h, respectively). In the experimental group, edema was detected
6 h following CCI in the left hippocampus (79.44±0.36%; P<0.05,
vs. sham), peaked at 24 h (81.28±0.20%; P<0.01, vs. sham) and
maintained a high level at 72 h (80.73±0.34%; P<0.01, vs. sham).
These results demonstrated the existence of acute hippocampal edema
following experimental CCI.
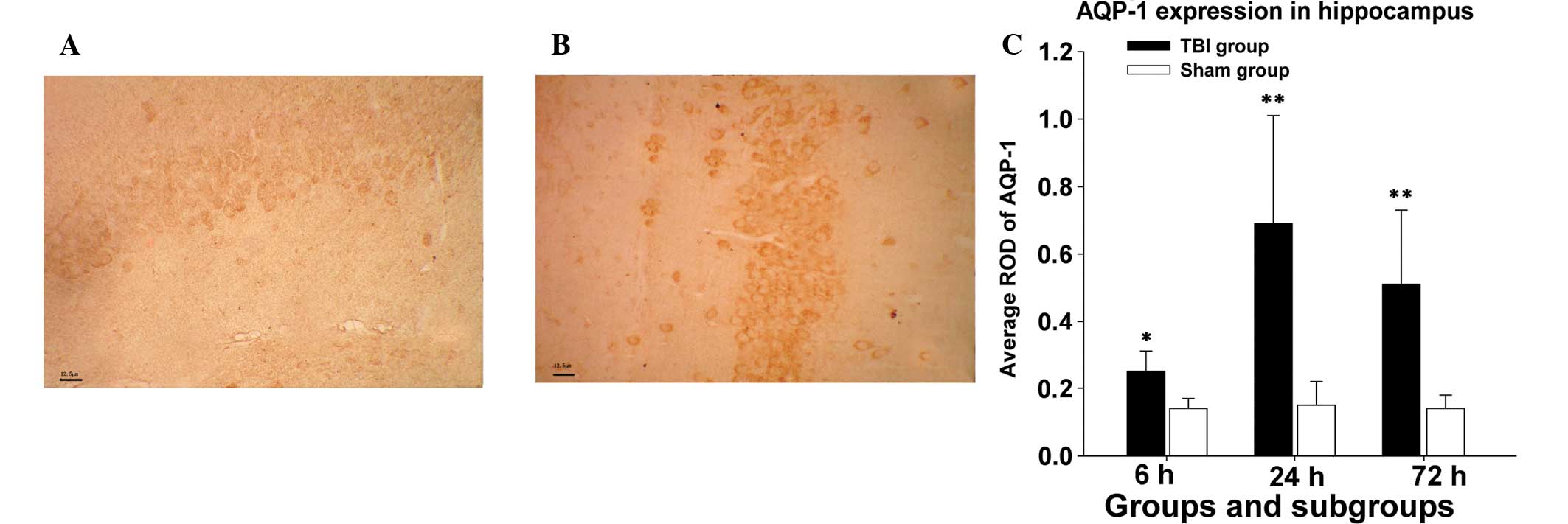
Outcomes of immunohistochemistry
Immunohistochemical staining for AQP-1 was evaluated
qualitatively in at least ten serial sections of each hippocampus
from a subgroup of experimental or sham groups and results of ten
separate measurements were expressed as the mean ± SD. The
intensity of the immunohistochemical reaction was expressed as
relative OD (ROD) of DAB brown reaction product and calculated
using Image-Pro Plus for Windows (Version 6.0; Media Cybernetics,
Bethesda, MD, USA) using the following formula where GL is the gray
level for the stained area (specimen, S) and unstained area
(background, BG) and blank (B) is the gray level measured following
removal of the slide from the light path: ROD =
ODS/ODBG =
log(GLB/GLS)/log(GLB/GLBG).
In the immunohistochemistry assay, AQP-1 expression
levels in mice hippocampi significantly increased 6 h following CCI
compared with those in the sham group at the same time (average,
0.25±0.06 vs. 0.14±0.03; P<0.05). AQP-1 expression in the
experimental group reached peak levels at 24 h following CCI
(0.69±0.32 vs. 0.15±0.07; P<0.01). Following 72 h CCI, AQP-1
expression marginally decreased, but remained markedly higher than
that in the sham group at the same time point (0.51±0.22 vs.
0.14±0.04; P<0.01). The difference in mean ROD of AQP-1 protein
levels in mice hippocampi between the experimental or sham group
was statistically significant at various times (6, 24 and 72 h), as
shown in Fig. 1.
Apoptosis detection in the hippocampus
following TBI
In the control group, the average apoptotic rate of
hippocampal cells was relatively constant and low at various times
(8.61±8.25, 7.55±9.54 and 10.17±6.08% at 6, 24 and 72 h following
sham surgery, respectively). In the experimental group, however,
the average apoptotic rate increased with prolonged post-injury
interval. A marked increase in TUNEL-positive cells was observed 6
h following injury (44.26±15.18%), which was significantly higher
than its counterpart in the sham group (P<0.01). The average
apoptotic rate of hippocampal cells in the experimental group
continued to rise at 24 h following injury (53.35±22.67%) and
reached 61.62±26.55% at 72 h following injury, which were
significantly higher values than those in corresponding subgroups
of the control group (P<0.01 for the two comparisons), as shown
in Fig. 2.
Quantitation of AQP-1 protein
expression
The present study investigated whether AQP-1
expression in BALB/c mice was altered following experimental TBI.
Western blot analysis showed one band at 28 kDa corresponding to
non-glycosylated AQP-1. There were significant differences in the
expression of AQP-1 between subgroups of the experimental and
control groups at various times (Fig.
3). The average AQP-1 expression level in the hippocampi of the
experimental group increased to 0.21±0.11 at 6 h, peaked
(0.46±0.19) at 24 h and marginally decreased to 0.35±0.17 at 72 h
following experimental TBI. Compared with those in the control
group at various times (0.14±0.02, 0.14±0.04 and 0.15±0.04, at 6,
24 and 72 h, respectively), the expression of AQP-1 protein was
markedly upregulated following TBI, which was in accordance with
the results of immunohistochemistry assay.
Discussion
Secondary insult of TBI includes complicated
biomolecular and physiological changes that destroy surviving cells
of the primary injury and cause delayed cell death in surrounding
or distant regions (2,26). Currently, post-injury hypoxia,
ischemia and edema have attracted increasing attention in TBI
research (1–4,7). The
contribution of cerebral edema to brain swelling in cases of TBI
remains a critical problem for which there is currently no
effective clinical treatment (7).
Notably, a previous study has demonstrated that the degree of brain
swelling assessed on the first computed tomography scan, obtained
soon after injury, correlates highly with mortality rate (e.g.,
compressed or obliterated mesencephalic cisterns vs. normal
cisterns, P<0.0002) (27),
indicating that therapy must be commenced as soon as possible to
avoid neurological deterioration or mortality (7). According to present hypotheses,
vasogenic edema may result from a failure of the blood-brain
barrier, while cytotoxic edema is the result of a biomolecular
injury and inflammatory cascade that causes loss of ionic
gradients, membrane dysfunction and cellular swelling (7,12,28).
As a result of traumatic impact or the subsequent attack of free
radicals, permeability of the blood-brain barrier or reflex
vasodilation of vessels increases and allows the plasma components
of blood to gain entrance to the extracellular space, indicating an
‘original’ vasogenic edema (1,2,7,13,14,29).
Additionally, ischemia and hypoxia, in the process of secondary
injury, may deplete cell energy stores and disable the
sodium-potassium ATPase. This reduces calcium exchange and,
thereby, results in cytotoxic cerebral edema (1,3).
Moreover, cellular swelling may, in turn, aggravate the cellular
dysfunction and cytotoxic edema (1,3).
Furthermore, previous studies of TBI have indicated that the
predominant type of edema in these injuries is cellular rather than
vasogenic (30,31). Therefore, the mechanisms of water
transport across the plasma membrane of brain cells are of
importance.
The identification of AQPs was acclaimed
enthusiastically, given that they may provide a mechanism for
massive water movement across the cell membrane (6,7,16–20).
Despite a common molecular structure, mammalian AQPs have been
subdivided into three functional groups according to permeability
characteristics: the AQPs (AQP-0, -1, -2, -4, -5 and -6), which are
permeable to water; the aquaglyceroporins (AQP-3, -7 and -8), which
are permeable to water and small nonpolar solutes; and the neutral
solute channels (AQP-9), which are permeable to water and specific
neutral solutes (7,18,32).
At present, AQPs are considered to contribute to cerebral edema
secondary to TBI, cerebral ischemia or hypoxia (6,16,18,33).
Although AQP-4 and -9 have been proven to be important in cerebral
edema following TBI or cerebral ischemia or hypoxia (6,7,16,18,33,34),
AQP-1, as the first AQP to be identified, was seldom investigated
in cerebral edema following TBI, despite there being evidence that
AQP-1 may participate in acute lung edema (35). In addition, the hippocampus is an
important organ that is extremely sensitive to ischemia, hypoxia
and edema, which may lead to hippocampal cell apoptosis and
subsequent cognitive, memory and learning impairments or epilepsy,
thereby profoundly affecting prognosis (8–14).
To the best of our knowledge, expression of AQP-1 in the
hippocampus following TBI has not yet been investigated. However,
studies have reported that upregulated AQP-1 expression following
acute hyponatremia correlates with necrotic cell mortality of the
hippocampal CA1 region, increasing water transport across the
blood-cerebrospinal fluid (CSF) barrier (19) or in vacuolized astrocytes in the
stratum radiatum of the CA1 region in an animal epilepsy model
(15). Thus, in the present study,
a series of assays was performed to develop mouse TBI models and
detect AQP-1 expression in the hippocampus following trauma and,
subsequently, to evaluate its possible effects in hippocampal
injuries correlated with TBI.
In this study, the mouse models were well
established and mimicked clinical manifestations of TBI.
Post-injury NSS confirmed that the majority of mice in the TBI
group were severely injured and attained the experimental
requirements. Acute hippocampal edema was also reproduced and
confirmed by evaluation of hippocampal tissue water content. In
situ apoptosis detection of hippocampal cells demonstrated a
progressive increase of apoptosis in the TBI group over time,
whereas the average apoptotic rate of hippocampal cells in the sham
group was maintained at a low and stable level. The differences
were statistically significantly (P<0.01 for all comparisons),
indicating an activation of hippocampal apoptosis following
experimental TBI, in which several pathophysiological processes may
be involved. The average apoptotic rate increased gradually in a
time-dependent manner, implying secondary or delayed cell mortality
or damage, resulting from physiological and biochemical changes
induced by the primary insult (26).
AQP-1 was formerly found to be expressed on
epithelial cells of the choroid plexus, with a role in CSF
production; however, more recently AQP-1 has been found to be
expressed in microvascular endothelial cells or astrocytes,
particularly in a number of pathological states, including brain
injury, subarachnoid hemorrhage or brain tumors, leading to edema
formation (16,21,26,36–38).
We hypothesize that AQP-1 is involved in TBI-associated cerebral
edema and this protein may be upregulated in response to a TBI
insult. Immunohistochemistry and western blot analysis
substantiated the upregulation of AQP-1 expression following TBI,
and the acute hippocampal edema was formed and evolved. Results of
the two assays were consistent, demonstrating enhanced AQP-1
expression levels at 6 h, maximum expression at 24 h and mildly
decreased (although remaining relatively high) expression at 72 h
following injury. These observations imply the involvement of AQP-1
in post-TBI pathophysiological and biochemical changes in the
hippocampus. Moreover, Kim et al (15) reported that AQP-1 overexpression
correlates with vacuolization of hippocampal CA1 astrocytes, which
may be responsible for subsequent apoptosis. Jablonski et al
(39) reported that AQP-1-mediated
water loss is important for the apoptotic volume decrease and
downstream apoptotic events and that the water permeability of the
plasma membrane may control the rate of apoptosis. Therefore, in
light of concurrent hippocampal edema and apoptosis exhibiting a
similar trend of development, it may be inferred that AQP-1
participates in edema formation and delayed cell mortality of the
hippocampus.
Currently, several studies have identified that
AQP-1 participates in the mediation of vasogenic edema (21,40);
furthermore, there is specific evidence to indicate that AQP-1
channels facilitate membrane CO2 permeability,
contributing to intracellular acidification (17,41).
Besides vasogenic edema, AQP-1-induced intracellular acidification
is likely to contribute to cytotoxic edema of the hippocampus in
that intracellular acidosis leads to ion transport dysfunction of
the cell membrane and subsequent cell swelling (17,42–45).
Additionally, vasogenic and cytotoxic edema may exacerbate cell
metabolic disorders and acidosis and, in turn, aggravate cerebral
edema (7,12,28).
Selective vulnerability disposes the hippocampus to a high
susceptibility to these pathophysiological attacks. As a result,
the post-TBI apoptotic cells in the hippocampus increased in a
time-dependent manner in the current study.
In conclusion, AQP-1 expression is upregulated in
the hippocampus of a mouse model following experimental TBI and may
exhibit differential roles in various responses, including post-TBI
hippocampal edema and apoptosis. Therefore, observations of the
present study indicate that AQP-1 may be a novel therapeutic target
to protect the hippocampus from secondary injury following TBI.
Acknowledgements
This study was supported by grants from the Chinese
National Natural Science Foundation of Youth Science Foundation
(grant nos. 81000565 and 81101917), the China Postdoctoral Science
Foundation (grant no. 2012M520831) and the Liaoning Provincial
Natural Science Foundation (grant nos. 201102095 and
2013021075).
References
|
1
|
Greve MW and Zink BJ: Pathophysiology of
traumatic brain injury. Mt Sinai J Med. 76:97–104. 2009. View Article : Google Scholar
|
|
2
|
Borgens RB and Liu-Snyder P: Understanding
secondary injury. Q Rev Biol. 87:89–127. 2012. View Article : Google Scholar
|
|
3
|
Werner C and Engelhard K: Pathophysiology
of traumatic brain injury. Br J Anaesth. 99:4–9. 2007. View Article : Google Scholar
|
|
4
|
Padayachy LC, Rohlwink U, Zwane E, Fieggen
G, Peter JC and Figaji AA: The frequency of cerebral
ischemia/hypoxia in pediatric severe traumatic brain injury. Childs
Nerv Syst. 28:1911–1918. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Walcott BP, Kahle KT and Simard JM: Novel
treatment targets for cerebral edema. Neurotherapeutics. 9:65–72.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adeva MM, Souto G, Donapetry C, Portals M,
Rodriguez A and Lamas D: Brain edema in diseases of different
etiology. Neurochem Int. 61:166–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marmarou A: A review of progress in
understanding the pathophysiology and treatment of brain edema.
Neurosurg Focus. 22:E12007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmidt-Kastner R and Freund TF: Selective
vulnerability of the hippocampus in brain ischemia. Neuroscience.
40:599–636. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Royo NC, Conte V, Saatman KE, et al:
Hippocampal vulnerability following traumatic brain injury: a
potential role for neurotrophin-4/5 in pyramidal cell
neuroprotection. Eur J Neurosci. 23:1089–1102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Deng P and Xu ZC: Contribution of Ih to
neuronal damage in the hippocampus after traumatic brain injury in
rats. J Neurotrauma. 28:1173–1183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nawashiro H, Shima K and Chigasaki H:
Selective vulnerability of hippocampal CA3 neurons to hypoxia after
mild concussion in the rat. Neurol Res. 17:455–460. 1995.PubMed/NCBI
|
|
12
|
Kawamata T, Katayama Y, Tsuji N and
Nishimoto H: Metabolic derangements in interstitial brain edema
with preserved blood flow: selective vulnerability of the
hippocampal CA3 region in rat hydrocephalus. Acta Neurochir Suppl.
86:545–547. 2003.PubMed/NCBI
|
|
13
|
Deng W, Aimone JB and Gage FH: New neurons
and new memories: how does adult hippocampal neurogenesis affect
learning and memory? Nat Rev Neurosci. 11:339–350. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eichenbaum H: Hippocampus: cognitive
processes and neural representations that underlie declarative
memory. Neuron. 44:109–120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JE, Ryu HJ, Yeo SI, et al:
Differential expressions of aquaporin subtypes in astroglia in the
hippocampus of chronic epileptic rats. Neuroscience. 163:781–789.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zelenina M: Regulation of brain
aquaporins. Neurochem Int. 57:468–488. 2010. View Article : Google Scholar
|
|
17
|
Tran ND, Kim S, Vincent HK, et al:
Aquaporin-1-mediated cerebral edema following traumatic brain
injury: effects of acidosis and corticosteroid administration. J
Neurosurg. 112:1095–1104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Badaut J, Lasbennes F, Magistretti PJ and
Regli L: Aquaporins in brain: distribution, physiology, and
pathophysiology. J Cereb Blood Flow Metab. 22:367–378. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim J and Jung Y: Increased aquaporin-1
and Na+-K+-2Cl− cotransporter 1
expression in choroid plexus leads to blood-cerebrospinal fluid
barrier disruption and necrosis of hippocampal CA1 cells in acute
rat models of hyponatremia. J Neurosci Res. 90:1437–1444. 2012.
|
|
20
|
Ameli PA, Madan M, Chigurupati S, Yu A,
Chan SL and Pattisapu JV: Effect of acetazolamide on aquaporin-1
and fluid flow in cultured choroid plexus. Acta Neurochir Suppl.
113:59–64. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suzuki R, Okuda M, Asai J, et al:
Astrocytes co-express aquaporin-1, -4, and vascular endothelial
growth factor in brain edema tissue associated with brain
contusion. Acta Neurochir Suppl. 96:398–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Constantini S, Trembovler V,
Weinstock M and Shohami E: An experimental model of closed head
injury in mice: pathophysiology, histopathology, and cognitive
deficits. J Neurotrauma. 13:557–568. 1996.PubMed/NCBI
|
|
23
|
Albert-Weißenberger C, Várrallyay C,
Raslan F, Kleinschnitz C and Sirén AL: An experimental protocol for
mimicking pathomechanisms of traumatic brain injury in mice. Exp
Transl Stroke Med. 4:12012.PubMed/NCBI
|
|
24
|
Shapira Y, Shohami E, Sidi A, Soffer D,
Freeman S and Cotev S: Experimental closed head injury in rats:
mechanical, pathophysiologic, and neurologic properties. Crit Care
Med. 16:258–265. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qiu B, Sun X, Zhang D, Wang Y, Tao J and
Ou S: TRAIL and paclitaxel synergize to kill U87 cells and
U87-derived stem-like cells in vitro. Int J Mol Sci.
13:9142–9156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stoica BA and Faden AI: Cell death
mechanisms and modulation in traumatic brain injury.
Neurotherapeutics. 7:3–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eisenberg HM, Gary HE Jr, Aldrich EF, et
al: Initial CT findings in 753 patients with severe head injury. A
report from the NIH Traumatic Coma Data Bank. J Neurosurg.
73:688–698. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Donkin JJ and Vink R: Mechanisms of
cerebral edema in traumatic brain injury: therapeutic developments.
Curr Opin Neurol. 23:293–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barzó P, Marmarou A, Fatouros P, Hayasaki
K and Corwin F: Contribution of vasogenic and cellular edema to
traumatic brain swelling measured by diffusion-weighted imaging. J
Neurosurg. 87:900–907. 1997.PubMed/NCBI
|
|
30
|
Ito J, Marmarou A, Barzó P, Fatouros P and
Corwin F: Characterization of edema by diffusion-weighted imaging
in experimental traumatic brain injury. J Neurosurg. 84:97–103.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Unterberg AW, Stover J, Kress B and
Kiening KL: Edema and brain trauma. Neuroscience. 129:1021–1029.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Verkman AS, Yang B, Song Y, Manley GT and
Ma T: Role of water channels in fluid transport studied by
phenotype analysis of aquaporin knockout mice. Exp Physiol. 85(Spec
No): 233S–241S. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Badaut J, Hirt L, Granziera C,
Bogousslavsky J, Magistretti PJ and Regli L: Astrocyte-specific
expression of aquaporin-9 in mouse brain is increased after
transient focal cerebral ischemia. J Cereb Blood Flow Metab.
21:477–482. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Badaut J: Aquaglyceroporin 9 in brain
pathologies. Neuroscience. 168:1047–1057. 2010. View Article : Google Scholar
|
|
35
|
Wang Q, Ishikawa T, Michiue T, Zhu BL,
Guan DW and Maeda H: Molecular pathology of pulmonary edema after
injury in forensic autopsy cases. Int J Legal Med. 126:875–882.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kobayashi H, Minami S, Itoh S, et al:
Aquaporin subtypes in rat cerebral microvessels. Neurosci Lett.
297:163–166. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Badaut J, Brunet JF, Grollimund L, et al:
Aquaporin 1 and aquaporin 4 expression in human brain after
subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir
Suppl. 86:495–498. 2003.PubMed/NCBI
|
|
38
|
Satoh J, Tabunoki H, Yamamura T, Arima K
and Konno H: Human astrocytes express aquaporin-1 and aquaporin-4
in vitro and in vivo. Neuropathology. 27:245–256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jablonski EM, Webb AN, McConnell NA, Riley
MC and Hughes FM Jr: Plasma membrane aquaporin activity can affect
the rate of apoptosis but is inhibited after apoptotic volume
decrease. Am J Physiol Cell Physiol. 286:C975–985. 2004.PubMed/NCBI
|
|
40
|
Kim J and Jung Y: Different expressions of
AQP1, AQP4, eNOS, and VEGF proteins in ischemic versus non-ischemic
cerebropathy in rats: potential roles of AQP1 and eNOS in
hydrocephalic and vasogenic edema formation. Anat Cell Biol.
44:295–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blank ME and Ehmke H: Aquaporin-1 and
HCO3(−)-Cl− transporter-mediated transport of
CO2 across the human erythrocyte membrane. J Physiol.
550:419–429. 2003.PubMed/NCBI
|
|
42
|
Staub F, Winkler A, Haberstok J, et al:
Swelling, intracellular acidosis, and damage of glial cells. Acta
Neurochir Suppl. 66:56–62. 1996.PubMed/NCBI
|
|
43
|
Ringel F, Plesnila N, Chang RC, Peters J,
Staub F and Baethmann A: Role of calcium ions in acidosis-induced
glial swelling. Acta Neurochir Suppl. 70:144–147. 1997.PubMed/NCBI
|
|
44
|
Plesnila N, Haberstok J, Peters J, Kolbl
I, Baethmann A and Staub F: Effect of lactacidosis on cell volume
and intracellular pH of astrocytes. J Neurotrauma. 16:831–841.
1999.PubMed/NCBI
|
|
45
|
Ringel F, Chang RC, Staub F, Baethmann A
and Plesnila N: Contribution of anion transporters to the
acidosis-induced swelling and intracellular acidification of glial
cells. J Neurochem. 75:125–132. 2000. View Article : Google Scholar : PubMed/NCBI
|