Introduction
Post-traumatic stress disorder (PTSD) is a severe
anxiety disorder that may develop following exposure to any threat
or injury that results in psychological trauma. Diagnostic symptoms
for PTSD include re-experiencing the original trauma through
flashbacks or nightmares, avoidance of stimuli associated with the
trauma and increased arousal. Single-prolonged stress (SPS), an
animal model of PTSD, has been extensively developed and employed
in the investigation of PTSD (1–3). The
three areas of the brain whose function may be altered in PTSD have
been identified as the prefrontal cortex, the amygdala and the
hippocampus, among which, the hippocampus is a key organ of the
limbic system involved in learning and memory, as well as being a
regulatory center for the stress response (4).
Alterations in brain neurochemistry have been linked
with neuropsychiatric disorders, including schizophrenia,
Alzheimer’s disease, depression and cognitive decline. Previous
studies have identified a positive association between vitamin D
signaling and cognitive function (5). Vitamin D may regulate
neurotransmission, neuroprotection and neuroimmunomodulation as a
neurosteroid hormone (5,6), in addition to its critical role in
calcium (Ca2+) and phosphorous regulation and skeletal
mineralization (7).
Hypovitaminosis D is associated with several neuropsychiatric
disorders, including dementia, Parkinson’s disease, multiple
sclerosis, epilepsy and schizophrenia. The action of vitamin D is
mediated by the vitamin D receptor (VDR), a ligand-activated
transcription factor (8). VDR is
considered to be a nuclear receptor that is ubiquitously expressed
in a wide variety of organs or tissues, including in the muscle,
adipose tissue, bone (9), cerebral
cortex and hippocampus (6,10). Genetic variance in the VDR gene
affects the susceptibility to age-related changes in cognitive
functioning and depressive symptoms (11). The absence of VDR has been
associated with neurodegenerative dementia, and VDR-knockout mouse
models have revealed that genetic ablation may cause anxiety and
motor disorders (12,13), indicating the essential role of VDR
in the brain (14).
Several lines of evidence indicate that an
alteration in neuronal Ca2+ homeostasis may be involved
in the neuroprotective actions of VDR. The appropriate
Ca2+ concentration is required for neuronal
excitability. Treatment with vitamin D decreases the density of
L-type voltage-sensitive Ca2+-channels (LVSCCs) and
protects against the excitotoxicity of the rat hippocampal cells
(15). Moreover, an increased
function of neuronal LVSCCs is strongly linked to impaired memory
and altered hippocampal synaptic plasticity in older rats (16), indicating that LVSCCs may also
contribute to the pathological memory changes during the
development of PTSD.
Since little is currently known about the
neurocognitive effects of VDR, it is of note to assay the
expression of this receptor in the hippocampus subjected to SPS,
where alterations are likely to occur during the development of
PTSD. Thus, in the present study, a rat model of PTSD was built
following the previously established SPS protocol (17,18),
and immunohistochemistry, reverse transcription-polymerase chain
reaction (RT-PCR) and western blotting analysis approaches were
used to identify the expression of VDR in the rat hippocampal
cells. In addition, the expression of LVSCC-A1C protein and mRNA
was examined, as well as the Ca2+ levels. To the best of
our knowledge, the present study is the first to directly assess
neuronal VDR and LVSCC-A1C activities in a rat model of PTSD.
Materials and methods
Animals
Young Sprague-Dawley male rats (6–7 weeks old)
weighing ~200 g were obtained from the Experimental Animal Center
of China Medical University (Shenyang, China). Animals were housed
singly under a 12-h light/dark cycle, with food and water freely
available. Following an adaptation period of 5–6 days, the
experimental procedures were undertaken. All procedures were
approved by the Institutional Animal Care and Use Committee (China
Medical University) and were in accordance with the National
Institutes of Health Guide for the care and use of laboratory
animals.
Experimental groups and the SPS
model
In total, 50 rats were randomly divided into five
groups; the control group and the SPS groups of 1, 3, 7 and 14
days, with 10 rats per group. The SPS model was created as
described previously, with slight modifications (17,18).
Briefly, rats were restrained for 2 h inside a disposable restraint
holder that was 58 mm in diameter and 150 mm in length. Next, they
were individually placed in a clear acrylic container of dimensions
600 mm × 400 mm × 500 mm, which was filled two-thirds with water at
24°C, and forced to swim for 20 min. Following a 15-min
recuperation, the animals were exposed to diethyl ether until loss
of consciousness and left undisturbed in their cages. The animals
were then randomly assigned to one of the four SPS groups.
Fixation and section preparation of the
hippocampus
Five rats from each group were anesthetized with
pentobarbital sodium (30 mg/kg intraperitoneally; China National
Medicines Corporation, Ltd., Shanghai, China) and perfused with 200
ml cold saline through the left ventricle, followed by perfusion
with 300 ml of 4% cold paraformaldehyde in phosphate buffer. The
whole brain was removed rapidly, dissected on ice and then fixed in
the same fixative solution for 10 h at 4°C. Following immersion in
a 20% sucrose solution for 24 h, the brain was sliced into 7-μm
coronal sections and stored at −70°C.
Immunohistochemical analysis of VDR
The sections were treated with 5% bovine serum
albumin and 0.3% Triton X-100 (Beyotime, Haimen, China) in
phosphate-buffered saline (PBS) for 30 min at room temperature for
blocking of non-specific staining, followed by incubation with
rabbit polyclonal antibody against VDR (Santa Cruz Biotechnology
Inc., Santa Cruz, CA, USA; 1:200) overnight at 4°C. Following
washing with PBS, the sections were incubated with anti-rabbit
immunoglobulin G horseradish peroxidase (HRP) antibody (ZSBio,
Beijing, China) for 0.5 h at 37°C. Finally, 3,3′-diaminobenzidine
was used as chromogen for 10 min until the brown coloring appeared.
Slices were then dehydrated and mounted with neutral gum. To assess
non-specific staining, a few sections in every experiment were
incubated in PBS without primary antibody.
Five slides were randomly selected from each group,
and on each slide, five visual fields were randomly selected
(magnification, ×200). The optical density (OD) of the positive
cells in each field was recorded to evaluate the average value. The
OD of the VDR-immunopositive cells was analyzed using a
MetaMorph/DPIO/BX41 morphology image analysis system (Olympus,
Tokyo, Japan).
Western blot analysis
The rats of each group were decapitated rapidly and
the hippocampi were dissected on ice. The samples were homogenized
with loading buffer containing 200 mM Tris-buffered saline, 4%
sodium dodecyl sulfate, 20% glycerol and 10% 2-mercaptoethanol, and
were denatured by boiling for 3 min. The protein fraction (30
μg/lane) extracted from each sample was separated by 12% (w/v)
gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and transferred to a 0.45-μm polyvinylidene fluoride (PVDF)
membrane (Millipore, Billerica, MA, USA). Following blocking with
5% (w/v) skimmed milk in 0.05% TBS with Tween-20 (TBST) at room
temperature for 2 h and incubation with a rabbit polyclonal
antibody against VDR (Santa Cruz; 1:200) or LVSCC-A1C rabbit
polyclonal antibody (Abcam, Cambridge, MA, USA; 1:200) overnight at
4°C, the membrane was incubated with anti-mouse IgG-HRP (Santa
Cruz; 1:5,000) secondary antibodies for another 2 h at room
temperature. Finally, the PVDF membrane was washed three times with
TBST prior to visualization using enhanced chemiluminescence
(Uscnlife, Wuhan, China). For each study, a representative
immunoblot from at least three independent experiments is
presented.
RT-PCR
Total mRNA was extracted from the hippocampus using
the TRIzol kit (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions. The primers were designed by Shenggong
Biotech Co., (Shanghai, China) according to the serial number from
Genbank, and are shown in Table I.
GAPDH mRNA used as an internal control was co-amplified with VDR or
LVSCC-A1C mRNA. The products were observed following
electrophoresis on a 1.2% agarose gel, and the density of each band
was analyzed with the Gel Image Analysis system (Tanon 2500R; Tanon
Science & Technology Co., Ltd., Shanghai, China). The levels of
VDR and LVSCC-A1C mRNA were determined by calculating the density
ratio of VDR or LVSCC-A1C mRNA to GAPDH mRNA.
 | Table IOligonucleotide sequences and product
sizes of the primers. |
Table I
Oligonucleotide sequences and product
sizes of the primers.
| Gene | Primer | Sequence | Product size,
bp |
|---|
| VDR | Forward
Reverse |
GTCTGCAGCGTGTTGGATAG
ATGACTCTACCCACGGCAAG | 157 |
| LVSCC-A1C | Forward
Reverse |
AATCTGACGGGAAAAAAGATGAA
TCCTGTCGACTCCTTAGTTAATCCT | 447 |
| GAPDH | Forward
Reverse |
ACGCCAGTAGACTCCACGAC
ATGACTCTACCCACGGCAAG | 178 |
Intracellular free Ca2+
assay
The rats of each group were decapitated rapidly and
the hippocampi were dissected on ice. A cell suspension of
106–107 cells/ml was achieved with a routine
method and loaded with 1 mmol/l fura-2-acetoxymethyl ester
(Beyotime) for 35 min, and then analyzed with a spectrofluorometer
(F-4500FL Fluorescence Spectrophotometer; Hitachi, Tokyo, Japan),
following the manufacturer’s instructions.
Statistical analysis
Data are presented as the mean ± standard deviation
and were analyzed with SPSS software (version 20.0; IBM, Armonk,
NY, USA). A one-way analysis of variance (ANOVA) with post hoc
Tukey’s test was used to determine statistical significance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Immunohistochemical analysis of VDR
expression
Following SPS stimulation, the hippocampi from the
treated and non-treated rats were analyzed with immunohistochemical
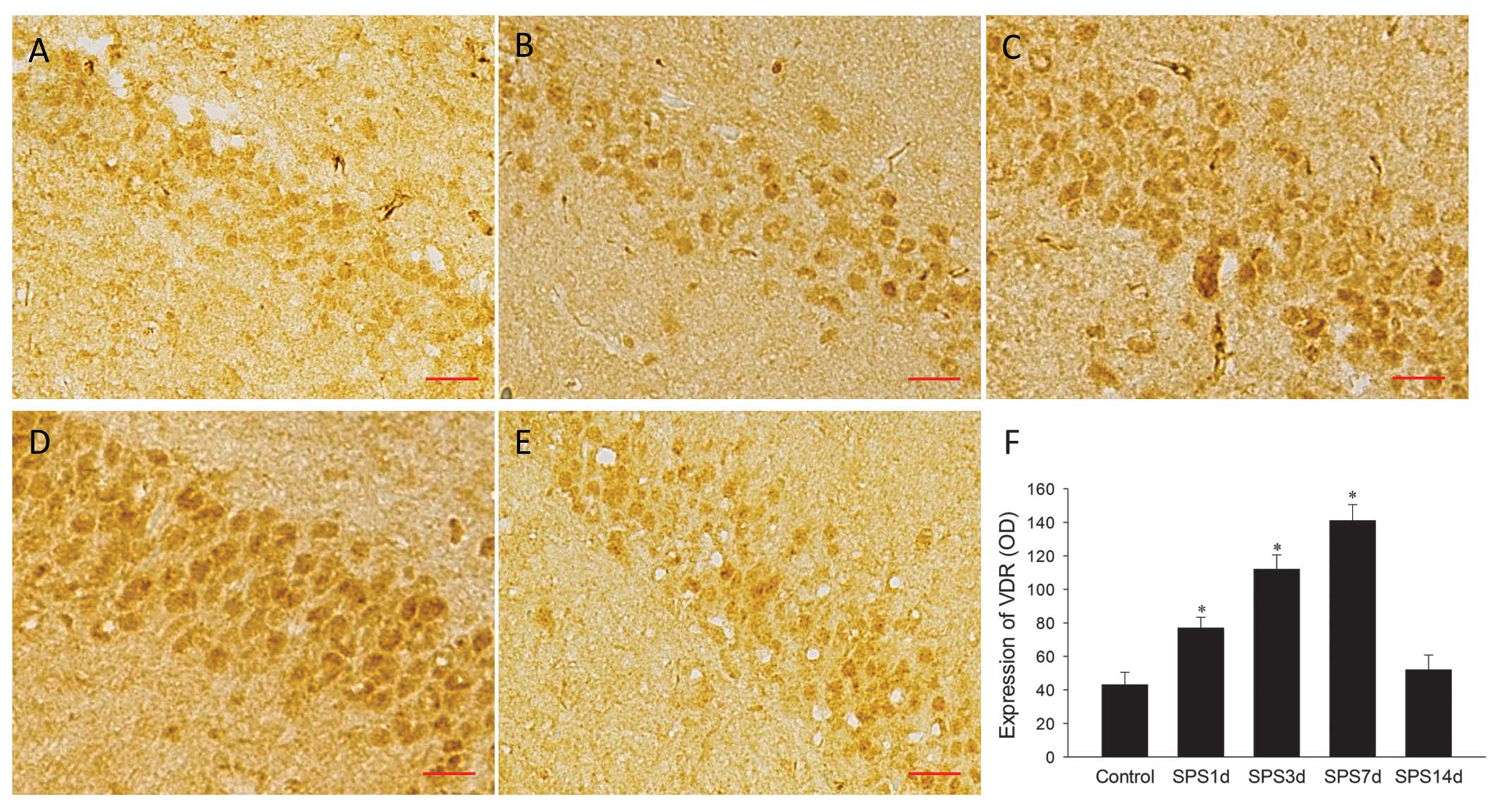
staining, as shown in Fig. 1A–E.
Positive immunohistochemical cells stained with the antibody
against VDR were brown, the majority revealing strong nuclear and
cytoplasmic staining; however, certain cells had extremely light
staining, indicating variations in the levels of VDR expression
among neurons. The evaluation of VDR expression by the mean ODs
indicated a significant change in the SPS 1, 3 and 7 day groups
compared with the control group (P<0.05; Fig. 1F). The peak of the increase was at
SPS 7 days. The immunoreactivity then decreased significantly to
its normal level at SPS 14 days. (P>0.05 vs. control).
Western blotting analysis of VDR and
LVSCC-A1C proteins
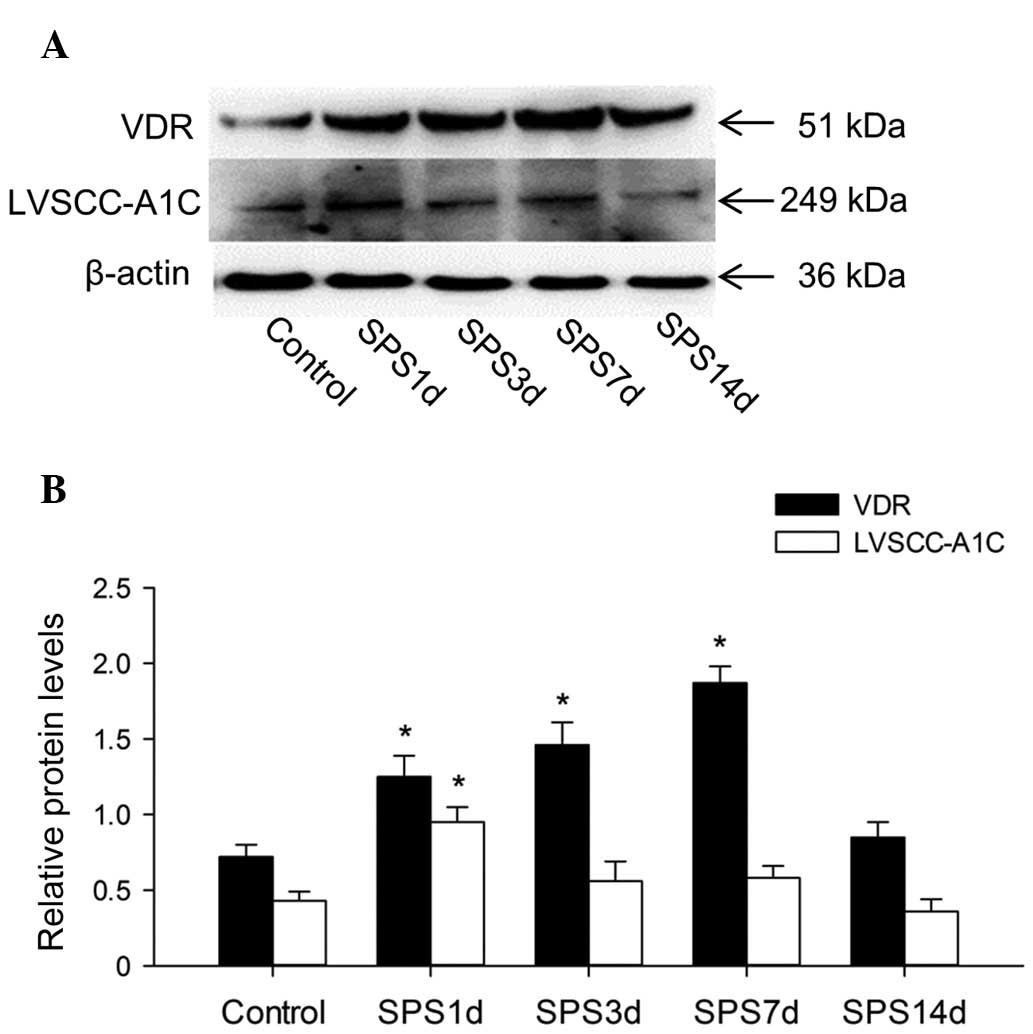
The VDR, LVSCC-A1C and β-actin proteins were
detected at 51, 249 and 36 kDa, respectively (Fig. 2A), and the mean values of the band
densities of the control group were set as 100%. The data were
expressed as normalized ODs. The OD value of the VDR bands had a
significant increase at 1, 3 and 7 days in the SPS groups compared
with the control group (P<0.05; Fig. 2B), while at 14 days it returned to
its normal value compared with the control group (P>0.05). The
OD value of LVSCC-A1C was upregulated at day 1 (P<0.05 vs.
control), and then had a significant reduction at 3, 7 and 14 days
(P>0.05 vs. control; Fig.
2B).
RT-PCR results
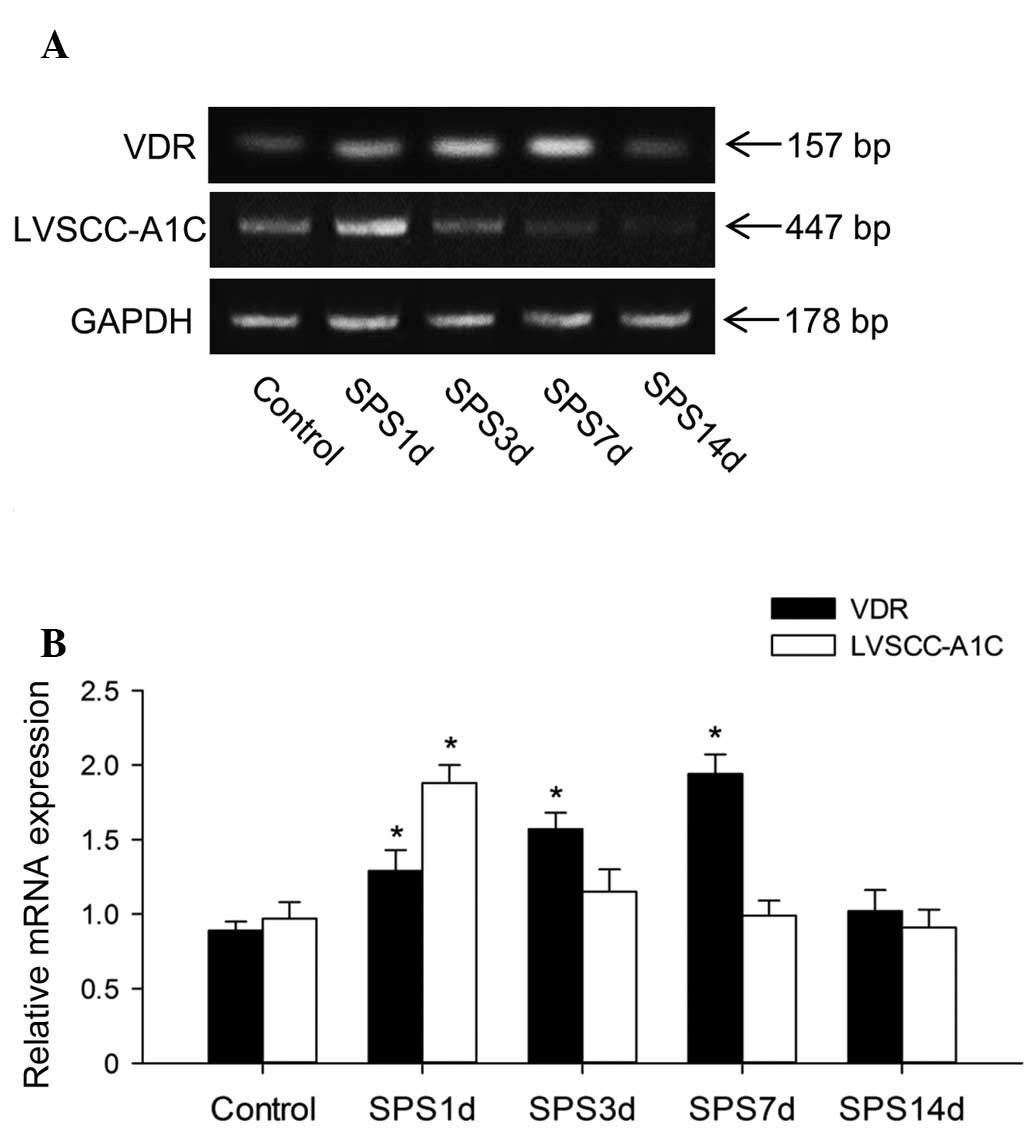
To further confirm the changes in VDR and LVSCC-A1C
expression caused by SPS exposure, RT-PCR analysis was performed
(Fig. 3A). The levels of VDR and
LVSCC-A1C mRNA were normalized with GAPDH mRNA. In the analysis of
VDR, the one-way ANOVA with post hoc analysis revealed that there
were significant differences between the SPS 1, 3 and 7 day groups
and the control group, respectively (P<0.05; Fig. 3B). Immediately following SPS, the
bands of the 1 day group demonstrated a significant upregulation of
the LVSCC-A1C mRNA level in the hippocampus compared with the
unexposed control group (P<0.05), which was then downregulated
to its normal level at 3 days (P>0.05).
Free Ca2+ concentration in the
hippocampus
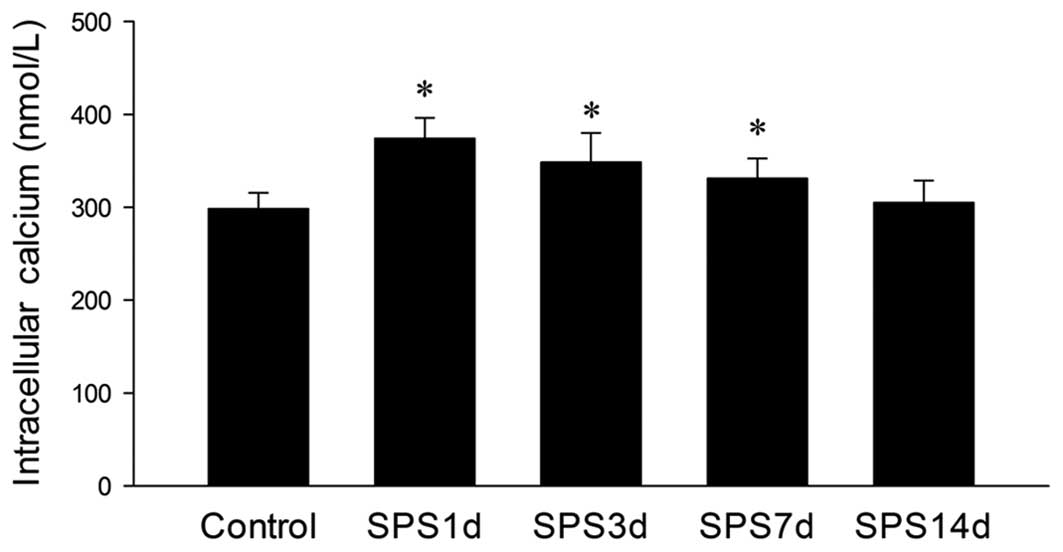
Following SPS exposure, the rats had an increased
intracellular free Ca2+ level in the hippocampal neurons
compared with the control group. The increase peaked 1 day
following exposure to SPS, and then decreased to normal levels at
14 days (Fig. 4).
Discussion
In this study, the detection of the free
Ca2+ content in the hippocampal neurons revealed
Ca2+ overload 1 day after SPS stimulation, which
gradually decreased to the normal levels at 14 days. Further
analysis of LVSCC-A1C, one of the main Ca2+ regulatory
proteins in the central nervous system (CNS), revealed that the
expression of LVSCC-A1C in the hippocampus markedly increased 1 day
after SPS stimulation, indicating that the LVSCC-A1C content
changed synchronously with the change of Ca2+
concentration. This occurred as the increased intracellular free
Ca2+ levels in the hippocampal neurons induced
overexpression of the LVSCC-A1C protein after SPS exposure, and the
increased LVSCC-A1C caused increased Ca2+ influx as a
positive feedback. VDR expression was also demonstrated to have
increased significantly at day 1, and reached its peak 7 days after
SPS stimulation, synchronously with the downregulation of
intracellular Ca2+ level 1 day after SPS. Notably, the
expression of LVSCC-A1C also reached its peak at day 1 and then
decreased significantly. Thus, it was postulated that the increased
VDR expression in the hippocampus may interact with LVSCC-mediated
Ca2+ dysregulation during the development of PTSD.
Therefore, VDR may also be important for modulating Ca2+
homeostasis, which is well recognized to be critical in
neuroprotection.
The presence of VDR in the CNS was first identified
in 1982 (19). There is now ample
evidence that VDR and its cascade enzymes are distributed in
different regions of the brain, and that the VDR signaling system
acts within the CNS as a neurosteroid with multiple actions
(20,21). However, evidence indicating a
correlation between VDR and stress is limited. It is unclear
whether VDR is involved in neurogenesis or Ca2+
homeostasis in response to stress. Little is known about whether
and how disturbing the function of the VDR affects the development
of post-traumatic stress disorder, since the regulators of VDR
expression in the CNS remains unknown (22). However in vitro evidence
indicates that there is cross-talk between the VDR and
glucocorticoid receptors in the hippocampus, and that vitamin D is
involved in neuronal differentiation and/or apoptosis in this
region (23). Mice lacking VDR
have exhibited substantial behavioral impairment and increased
anxiety (24).
It is now better recognized that vitamin D, the
ligand of VDR, exhibits a role in the nervous system. In a number
of studies, it has been indicated that vitamin D in the brain
regulates neurotrophic factor expression, oxidative stress
mechanisms and Ca2+ homeostasis (25–31).
Vitamin D may affect neuronal plasticity processes and increase
neurite outgrowth when added to cultured hippocampal cells
(28). It has been revealed that
vitamin D upregulates the expression of microtubule-associated
protein-2 and growth-associated protein-43 in cultured cortical
neurons (32). Maternal
hypovitaminosis D decreases the expression of proteins involved in
cytoskeleton maintenance, including neurofilaments, tubulin, actin
and glial fibrillary acidic protein (33,34).
More recently, studies have indicated a potential beneficial role
of vitamin D in cognitive function and neuroprotective effects
beyond classical mineral homeostasis. Defects in the vitamin D
signaling system have been associated with various neuropsychiatric
disorders (35–37). Human vitamin D deficiency may
result in an active mood disorder and worse cognitive functioning
(38,39). In animals, it has been demonstrated
that prenatal vitamin D deficiency resulted in alterations in brain
morphology, learning and memory (40,41).
In addition, mice lacking a functional VDR gene exhibited
anxiety-like behavior, indicating that vitamin D may affect
cognitive functioning and the prevalence of depressive symptoms
(13).
The vitamin D signaling system is essential in
overall Ca2+ homeostasis. Acute exposure to
1,25(OH)2D3 increases the mean open time and
plasma membrane Ca2+ permeability of the LVSCC in the
short term, thereby easing the requirement for Ca2+
influx (42). VSCCs mediate the
influx of Ca2+ in response to membrane depolarization,
and regulate intracellular functions, including
excitation-secretion, gene transcription, neurotransmitter release
and cell differentiation. The neuroprotective effects of vitamin D
appear to be exerted via the regulation of Ca2+
homeostasis and the synthesis of neurotrophins, which support the
survival of existing neurons and the growth and differentiation of
new neurons (26,27,30,43).
Previous studies have indicated that Ca2+ dysregulation
is involved in the aging brain and in Alzheimer’s disease, giving
rise to the ‘Ca2+ hypothesis of brain aging and
dementia’ (44). Ca2+
overload and the dysregulation of Ca2+ signaling in the
nerve cells in PTSD patients have been shown to increase the
cytotoxicity of the brain neurons (45). The detrimental effects of excessive
Ca2+ on memory formation and cognitive functioning are
widely acknowledged (46–49). Administration of vitamin D or its
metabolites may decrease neuronal death in rat hippocampal
cultures, elicited by Ca2+-mediated neurotoxicity
through the downregulation of LVSCC and increased VDR levels
(26,32).
To date, the pathogenesis of PTSD is far from
definite. PTSD may result from a series of biochemical and
physiological abnormalities in the brain, which leads to
dysfunction of the hippocampus. Collectively, the findings of this
study provide a novel perspective on the pathogenesis of PTSD.
Further evidence of the actions of VDR in the hippocampus may
indicate its potential effects in a wide range of neuropsychiatric
disorders.
Acknowledgements
This study was supported by the Research Fund for
Social Development of Science and Technology of Liaoning Province,
China (grant no. 2012225021).
References
|
1
|
Liberzon I and Young EA: Effects of stress
and glucocorticoids on CNS oxytocin receptor binding.
Psychoneuroendocrinology. 22:411–422. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khan S and Liberzon I: Topiramate
attenuates exaggerated acoustic startle in an animal model of PTSD.
Psychopharmacology (Berl). 172:225–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iwamoto Y, Morinobu S, Takahashi T and
Yamawaki S: Single prolonged stress increases contextual freezing
and the expression of glycine transporter 1 and vesicle-associated
membrane protein 2 mRNA in the hippocampus of rats. Prog
Neuropsychopharmacol Biol Psychiatry. 31:642–651. 2007. View Article : Google Scholar
|
|
4
|
Joëls M: Functional actions of
corticosteroids in the hippocampus. Eur J Pharmacol. 583:312–321.
2008.
|
|
5
|
Buell JS and Dawson-Hughes B: Vitamin D
and neurocognitive dysfunction: preventing ‘D’ecline? Mol Aspects
Med. 29:415–422. 2008.PubMed/NCBI
|
|
6
|
Kalueff AV and Tuohimaa P: Neurosteroid
hormone vitamin D and its utility in clinical nutrition. Curr Opin
Clin Nutr Metab Care. 10:12–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartoccini E, Marini F, Damaskopoulou E,
et al: Nuclear lipid microdomains regulate nuclear vitamin D3
uptake and influence embryonic hippocampal cell differentiation.
Mol Biol Cell. 22:3022–3031. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pike JW and Meyer MB: The vitamin D
receptor: new paradigms for the regulation of gene expression by
1,25-dihydroxyvitamin D(3). Endocrinol Metab Clin North Am.
39:255–269. 2010. View Article : Google Scholar
|
|
9
|
Freeman MR, Cinar B, Kim J, et al: Transit
of hormonal and EGF receptor-dependent signals through
cholesterol-rich membranes. Steroids. 72:210–217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marini F, Bartoccini E, Cascianelli G, et
al: Effect of 1alpha,25-dihydroxyvitamin D3 in embryonic
hippocampal cells. Hippocampus. 20:696–705. 2010.PubMed/NCBI
|
|
11
|
Kuningas M, Mooijaart SP, Jolles J,
Slagboom PE, Westendorp RG and van Heemst D: VDR gene variants
associate with cognitive function and depressive symptoms in old
age. Neurobiol Aging. 30:466–473. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burne TH, Johnston AN, McGrath JJ and
Mackay-Sim A: Swimming behaviour and post-swimming activity in
Vitamin D receptor knockout mice. Brain Res Bull. 69:74–78. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kalueff AV, Lou YR, Laaksi I and Tuohimaa
P: Increased anxiety in mice lacking vitamin D receptor gene.
Neuroreport. 15:1271–1274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sutherland MK, Somerville MJ, Yoong LK,
Bergeron C, Haussler MR and McLachlan DR: Reduction of vitamin D
hormone receptor mRNA levels in Alzheimer as compared to Huntington
hippocampus: correlation with calbindin-28k mRNA levels. Brain Res
Mol Brain Res. 13:239–250. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Langub MC, Herman JP, Malluche HH and
Koszewski NJ: Evidence of functional vitamin D receptors in rat
hippocampus. Neuroscience. 104:49–56. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thibault O, Pancani T, Landfield PW and
Norris CM: Reduction in neuronal L-type calcium channel activity in
a double knock-in mouse model of Alzheimer’s disease. Biochim
Biophys Acta. 1822:546–549. 2012.PubMed/NCBI
|
|
17
|
Takahashi T, Morinobu S, Iwamoto Y and
Yamawaki S: Effect of paroxetine on enhanced contextual fear
induced by single prolonged stress in rats. Psychopharmacology
(Berl). 189:165–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liberzon I, Krstov M and Young EA:
Stress-restress: effects on ACTH and fast feedback.
Psychoneuroendocrinology. 22:443–453. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stumpf WE, Sar M, Clark SA and DeLuca HF:
Brain target sites for 1,25-dihydroxyvitamin D3. Science.
215:1403–1405. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eyles DW, Smith S, Kinobe R, Hewison M and
McGrath JJ: Distribution of the vitamin D receptor and 1
alpha-hydroxylase in human brain. J Chem Neuroanat. 29:21–30. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kiraly SJ, Kiraly MA, Hawe RD and Makhani
N: Vitamin D as a neuroactive substance: review. Scientific World
Journal. 6:125–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oudshoorn C, Mattace-Raso FU, van der
Velde N, Colin EM and van der Cammen TJ: Higher serum vitamin D3
levels are associated with better cognitive test performance in
patients with Alzheimer’s disease. Dement Geriatr Cogn Disord.
25:539–543. 2008.PubMed/NCBI
|
|
23
|
Obradovic D, Gronemeyer H, Lutz B and Rein
T: Cross-talk of vitamin D and glucocorticoids in hippocampal
cells. J Neurochem. 96:500–509. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Minasyan A, Keisala T, Lou YR, Kalueff AV
and Tuohimaa P: Neophobia, sensory and cognitive functions, and
hedonic responses in vitamin D receptor mutant mice. J Steroid
Biochem Mol Biol. 104:274–280. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bouillon R, Carmeliet G, Daci E, Segaert S
and Verstuyf A: Vitamin D metabolism and action. Osteoporos Int.
8(Suppl 2): S13–S19. 1998. View Article : Google Scholar
|
|
26
|
Brewer LD, Thibault V, Chen KC, Langub MC,
Landfield PW and Porter NM: Vitamin D hormone confers
neuroprotection in parallel with downregulation of L-type calcium
channel expression in hippocampal neurons. J Neurosci. 21:98–108.
2001.PubMed/NCBI
|
|
27
|
Brewer LD, Porter NM, Kerr DS, Landfield
PW and Thibault O: Chronic 1alpha,25-(OH)2 vitamin D3 treatment
reduces Ca2+-mediated hippocampal biomarkers of aging.
Cell Calcium. 40:277–286. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brown J, Bianco JI, McGrath JJ and Eyles
DW: 1,25-dihydroxyvitamin D3 induces nerve growth factor, promotes
neurite outgrowth and inhibits mitosis in embryonic rat hippocampal
neurons. Neurosci Lett. 343:139–143. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cornet A, Baudet C, Neveu I, Baron-Van
Evercooren A, Brachet P and Naveilhan P: 1,25-Dihydroxyvitamin D3
regulates the expression of VDR and NGF gene in Schwann cells in
vitro. J Neurosci Res. 53:742–746. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Neveu I, Naveilhan P, Jehan F, et al:
1,25-dihydroxyvitamin D3 regulates the synthesis of nerve growth
factor in primary cultures of glial cells. Brain Res Mol Brain Res.
24:70–76. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dursun E, Gezen-Ak D and Yilmazer S: A
novel perspective for Alzheimer’s disease: vitamin D receptor
suppression by amyloid-β and preventing the amyloid-β induced
alterations by vitamin D in cortical neurons. J Alzheimers Dis.
23:207–219. 2011.
|
|
32
|
Taniura H, Ito M, Sanada N, et al: Chronic
vitamin D3 treatment protects against neurotoxicity by glutamate in
association with upregulation of vitamin D receptor mRNA expression
in cultured rat cortical neurons. J Neurosci Res. 83:1179–1189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Almeras L, Eyles D, Benech P, et al:
Developmental vitamin D deficiency alters brain protein expression
in the adult rat: implications for neuropsychiatric disorders.
Proteomics. 7:769–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eyles D, Almeras L, Benech P, et al:
Developmental vitamin D deficiency alters the expression of genes
encoding mitochondrial, cytoskeletal and synaptic proteins in the
adult rat brain. J Steroid Biochem Mol Biol. 103:538–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cantorna MT, Hayes CE and DeLuca HF:
1,25-Dihydroxyvitamin D3 reversibly blocks the progression of
relapsing encephalomyelitis, a model of multiple sclerosis. Proc
Natl Acad Sci USA. 93:7861–7864. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Garcion E, Wion-Barbot N, Montero-Menei
CN, Berger F and Wion D: New clues about vitamin D functions in the
nervous system. Trends Endocrinol Metab. 13:100–105. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lansdowne AT and Provost SC: Vitamin D3
enhances mood in healthy subjects during winter. Psychopharmacology
(Berl). 135:319–323. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Przybelski RJ and Binkley NC: Is vitamin D
important for preserving cognition? A positive correlation of serum
25-hydroxyvitamin D concentration with cognitive function. Arch
Biochem Biophys. 460:202–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wilkins CH, Sheline YI, Roe CM, Birge SJ
and Morris JC: Vitamin D deficiency is associated with low mood and
worse cognitive performance in older adults. Am J Geriatr
Psychiatry. 14:1032–1040. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eyles D, Brown J, Mackay-Sim A, McGrath J
and Feron F: Vitamin D3 and brain development. Neuroscience.
118:641–653. 2003. View Article : Google Scholar
|
|
41
|
Becker A, Eyles DW, McGrath JJ and
Grecksch G: Transient prenatal vitamin D deficiency is associated
with subtle alterations in learning and memory functions in adult
rats. Behav Brain Res. 161:306–312. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bergh JJ, Shao Y, Puente E, Duncan RL and
Farach-Carson MC: Osteoblast Ca(2+) permeability and
voltage-sensitive Ca(2+) channel expression is temporally regulated
by 1,25-dihydroxyvitamin D(3). Am J Physiol Cell Physiol.
290:C822–C831. 2006.
|
|
43
|
Naveilhan P, Neveu I, Wion D and Brachet
P: 1,25-Dihydroxyvitamin D3, an inducer of glial cell line-derived
neurotrophic factor. Neuroreport. 7:2171–2175. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Thibault O, Gant JC and Landfield PW:
Expansion of the calcium hypothesis of brain aging and Alzheimer’s
disease: minding the store. Aging Cell. 6:307–317. 2007.PubMed/NCBI
|
|
45
|
Xiao B, Yu B, Wang HT, Han F and Shi YX:
Single-prolonged stress induces apoptosis by activating cytochrome
C/caspase-9 pathway in a rat model of post-traumatic stress
disorder. Cell Mol Neurobiol. 31:37–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sattler R and Tymianski M: Molecular
mechanisms of calcium-dependent excitotoxicity. J Mol Med (Berl).
78:3–13. 2000. View Article : Google Scholar
|
|
47
|
Arundine M and Tymianski M: Molecular
mechanisms of calcium-dependent neurodegeneration in
excitotoxicity. Cell Calcium. 34:325–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thibault O, Hadley R and Landfield PW:
Elevated postsynaptic [Ca2+]i and L-type calcium channel activity
in aged hippocampal neurons: relationship to impaired synaptic
plasticity. J Neurosci. 21:9744–9756. 2001.
|
|
49
|
Veng LM, Mesches MH and Browning MD:
Age-related working memory impairment is correlated with increases
in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1
of the hippocampus and both are ameliorated by chronic nimodipine
treatment. Brain Res Mol Brain Res. 110:193–202. 2003. View Article : Google Scholar
|