Introduction
Osteonecrosis (ON) is one of the most serious
complications induced by high doses and/or long-term administration
of glucocorticoids (GCs). Several mechanisms have been associated
with the pathogenesis of ON, including intraosseous hypertension,
oxidation injury, apoptosis, hypercoagulability and lipid
metabolism disorders (1–5). However, the precise mechanism
underlying the pathogenesis of steroid-induced ON is yet to be
elucidated and until recently, effective prophylactic therapies
have not been available.
The renin-angiotensin system (RAS) is classically
known to be a circulating endocrine system regulating blood
pressure and electrolyte homeostasis. The main effector peptide in
RAS is angiotensin (Ang) II, which is formed from Ang I by
angiotensin-converting enzyme (ACE). Ang II exerts its biological
effects through binding to specific angiotensin receptors,
primarily the Ang II type 1 (AT1) receptor. In addition
to this classical systemic RAS, additional local tissue-specific
RASs have been identified in various organs and tissues, including
the heart, kidney, bone marrow, blood vessels and fat tissues.
Moreover, the local RAS has been identified to have an important
role in local organ regulation (6).
The local RAS has been shown to exist in bone tissue
(7,8) and its activation in bone tissue has
been found to induce metabolic bone disorders (9–11).
Furthermore, Ang II-induced signaling in vascular and endothelial
cells promotes reactive oxygen species (ROS) production, platelet
activation, inflammation and altered vasoreactivity, all of which
impair bone microcirculation (12).
Previous studies have shown that GCs stimulate ACE
expression in bovine aorta endothelial cells, rat cardiac
fibroblasts and vascular smooth muscle cells (13–15).
Furthermore, Sato et al (16) showed that GCs upregulate the
expression of the AT1 receptor in vascular smooth muscle
cells. However, little is known about the role of GCs in the
regulation of the RAS in the bone.
In the present study, it was hypothesized that GCs
may activate the local bone RAS and that this activation may be
involved in the pathogenesis of steroid-induced ON. Therefore, this
study investigated the effect of steroid-induced ON on the
expression of Ang II, ACE and AT1 and Ang II type 2
(AT2) receptors in adult female Japanese white
rabbits.
Materials and methods
Animals
The experimental protocol was approved by the
institutional animal use and care review board of Xi’an Jiaotong
University (Xi’an, China). Fifty-five adult, female Japanese white
rabbits (weight, 3.3–4.2 kg; age, 30–32 weeks; Animal Center of
Xi’an Jiaotong University) were investigated. All rabbits were
housed at the Animal Center of Xi’an Jiaotong University and
maintained on a standard diet and water.
Forty-five rabbits were injected once with 20 mg/kg
body weight methylprednisolone acetate (MPA; Pfizer, Inc.,
Brussels, Belgium) into the right gluteal muscle, and were then
divided into three groups (A, B and C) consisting of 15 rabbits per
group. The rabbits in groups A, B and C were sacrificed by overdose
of anesthesia at 1, 2 and 3 weeks subsequent to MPA administration,
respectively. The control group (group N) consisted of 10 rabbits,
which were maintained under the same conditions as the treatment
groups, but were not injected with MPA (17). Immediately following sacrifice, one
half of each femoral head was isolated and fixed in 10% neutral
buffered formalin, decalcified using 13% EDTA and embedded in
paraffin. The other half of each femoral head was frozen and stored
at −80°C for additional examinations.
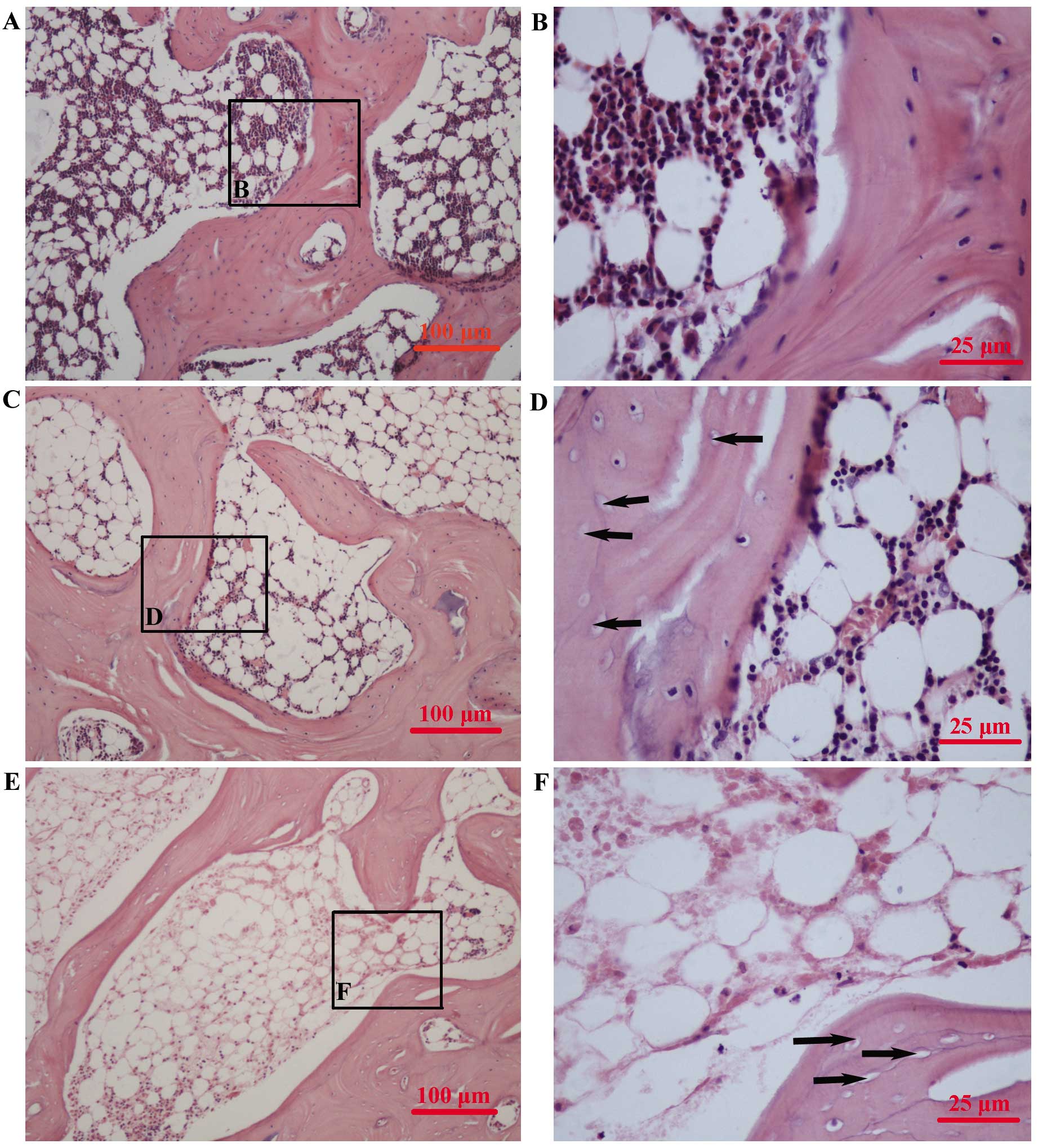
Assessment of ON
One 4-μm thick section of each femoral head was cut
in the coronal plane and stained with hematoxylin and eosin. The
presence or absence of ON was determined in whole areas of two
sections for each rabbit. The sections were examined using light
microscopy (Nikon YS100; Nikon Corporation, Toyko, Japan) by two
blinded pathologists. ON was identified based on the presence of
empty lacunae or pyknotic osteocyte nuclei in the bone trabeculae,
as well as the presence of necrosis in the surrounding bone marrow
or fat cells. Empty lacunae in the bone trabeculae, but without
bone marrow or fat cell necrosis was not classified as ON (Fig. 1). Rabbits were considered to have
ON based on the identification of ON in at least one of the two
sections analyzed. The incidence of ON was calculated as the ratio
of the number of rabbits with ON to the total number of rabbits
(17,18).
Immunohistochemistry
Immunohistochemistry was performed using one 4-μm
thick section of each femoral head in order to assess the presence
of AT1 receptors and ACE using specific antibodies
according to the manufacturer’s instructions. Briefly, subsequent
to deparaffinization, sections were treated with 3% hydrogen
peroxide for 20 min to inhibit endogenous peroxidase activity.
Antigen retrieval was then performed using 0.01 M citrate buffer
(pH 6.0) at 80°C for 10 min. Sections were preincubated with normal
goat serum (Biosynthesis Biotechnology Co. Ltd., Beijing, China)
for 30 min at room temperature, prior to incubation at 4°C
overnight with mouse anti-rabbit ACE (ab11734; Abcam PLC,
Cambridge, MA, USA) and AT1 receptor (ab9391; Abcam PLC)
monoclonal antibodies, diluted 1:20 and 1:50 in phosphate-buffered
saline, respectively. Sections were then incubated with secondary
goat anti-mouse antibodies (Biosynthesis Biotechnology Co. Ltd.)
and with horseradish peroxidase (HRP)-labeled streptavidin
(Biosynthesis Biotechnology Co. Ltd.). The final reaction product
was visualized using diaminobenzidine. Images were captured using
the QWin550CW Image Acquiring and Analysis system (Leica
Microsystems, Wetzlar, Germany). Heart tissue was used as a
positive control and showed positive brown staining. Sections
without primary antibody-treatment were used as negative
controls.
The intensity of AT1 receptor and ACE
immunostaining in groups N, A, B and C were quantitatively analyzed
using the analysis software Image-Pro Plus (Media Cybernetics,
Baltimore, MD, USA). One section was obtained from each rabbit and
10 images were captured from each section, which were analyzed for
positive staining at a magnification of ×400. The total area of
each analyzed section was the same. Integrated optical density
(IOD) was assessed, in which ‘integrated’ refers to the sum of all
the pixel intensity or density values in a given image. The IOD
values obtained from the 10 images in each section were averaged
and compared with the averaged IOD values of each section.
Western blot analysis
Six femoral heads were selected randomly from each
group for western blot analysis. Total protein was isolated by
homogenizing the femoral head using radioimmunoprecipitation assay
buffer (RIPA) buffer. The concentration of total protein was
quantified using the bicinchoninic acid (BCA) protein assay reagent
(Pierce™, Rockford, IL, USA). Laemmli buffer (5X) was added to each
sample to a final concentration of 1X, and 20 μl of each
preparation was loaded onto 5 and 10% SDS polyacrylamide gels.
SDS-PAGE was performed using a constant voltage of 90 V for 100
min. Following electrophoresis, proteins were transferred onto 0.45
μm nitrocellulose and polyvinylidene fluoride membranes
(Hybond-ECL; Amersham Pharmacia Biotechnology Inc., Piscataway, NJ,
USA) and blocked with 3% bovine serum albumin at room temperature
for 2 h. Membranes were incubated overnight at 4°C with
anti-AT1 receptor and -ACE primary antibodies (Abcam
PLC) diluted 1:400 and 1:100, respectively. Membranes were then
incubated with HRP-labeled goat anti-mouse secondary antibodies
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Immunoreactive proteins were visualized on a film using an enhanced
chemiluminescence kit (NEN Life Science Products Inc., Boston, MA,
USA). Relative protein expression was determined using image
analysis software (Media Cybernetics). β-actin was detected using a
mouse monoclonal anti-actin antibody (1:3,000; Santa Cruz
Biotechnology, Inc.) and was used as an internal control.
Analysis of ACE activity in the serum and
bone
Prior to sacrifice, blood was collected from all
rabbits without anticoagulant and was stored on ice. Blood was then
centrifuged at 1,848 × g for 10 min. The serum was obtained and
stored at −80°C until required for the ACE assay. A total of 200 mg
bone tissue was obtained from each rabbit and homogenized in
ice-cold Tris-HCl buffer solution (1 ml/100 mg sample wet weight).
The buffer solution consisted of 20 mM Tris-HCl (pH 8.3), 5 mM
Mg(CH3COO)2, 30 mM KCl, 250 mM sucrose and 0.5% Nonidet
P-40. The homogenized samples were centrifuged for 30 min at 11,300
× g at 4°C. The protein concentration in the supernatant was
quantified using the BCA protein assay reagent (Pierce). The
supernatant was stored at −80°C until required for the ACE
assay.
ACE activity in the serum and the supernatant was
determined by analyzing the production rate of hippuric acid from
the synthetic tripeptide substrate hippuryl-L-histidyl-L-leucine
(HHL) as described previously (19). The serum and the supernatant were
incubated with the substrate, HHL, and the hippuric acid
concentration was assessed using ultra violet absorbance at 228 nm.
ACE activity was expressed as nmol/min/mg protein or per ml serum.
All analyses were performed in duplicate.
Analysis of Ang II concentration in the
plasma and bone
Prior to sacrifice, blood was collected from all
rabbits with a mixture of protease inhibitors (0.30 M EDTA, 0.32 M
dimercaprol dimercaptopropanol and 0.34 M 8-sulfhydryl quinoline
sulfate) and stored on ice. Blood was then centrifuged at 943 × g
for 7 min. The plasma was obtained and stored at −80°C until
required for the Ang II assay. A total of 100 mg bone tissue was
obtained from each rabbit and homogenized and extracted in lysis
buffer containing 10 mM Tris, pH 7.5, 10 mM NaCl, 0.1 mM EDTA, 0.5%
Triton X-100, 0.02% NaN3 and 0.2 mM phenylmethylsulfonyl
fluoride protease inhibitor cocktail. The homogenized samples were
then centrifuged for 30 min at 11,300 × g at 4°C. The protein
concentration in the supernatant was quantified using the BCA
protein assay reagent (Pierce). The supernatant was stored at −80°C
until required for the Ang II assay. The concentration of Ang II
was measured using radioimmunoassay (RIA) (20) with a commercial RIA kit (Beifang,
Tianjin, China). The concentration of Ang II was expressed as pg/mg
protein or pg/ml plasma. All analyses were performed in
duplicate.
Quantitative polymerase chain reaction
(qPCR) analysis
Total RNA was isolated by homogenizing the femoral
heads using the TRIzol® protocol. cDNA was synthesized
using the RevertAid™ First Strand cDNA Synthesis kit (Fermentas,
Burlington, ON, Canada) according to the manufacturer’s
instructions. Samples were analyzed using SYBR-Green®
PCR Master mix (DRR820S; Takara Bio, Inc., Shiga, Japan) and an ABI
7300 Real-Time PCR system (Bio-Rad Laboratories, Hercules, CA,
USA). The sequences of the primers used for qPCR were as follows:
Forward: 5′-TGTAGCCAAAGTCACCTGCATC-3′ and reverse:
5′-ACTCGTAATGGAAAGCACAAACC-3′ for the AT1 receptor;
forward: 5′-ATAAGCCATCA GATAAGCAG TTAG-3′ and reverse:
5′-GAGGAAGAGTAGCCACAAGG-3′ for the AT2 receptor;
forward: 5′-GGA GCATTACCAA GGAGAACTAC-3′, and reverse: 5′-AAC
TGGAACTGGATG ATGAAGC-3′ for ACE; and forward: 5′-GTGCGGGACATC
AAGGAGA-3′ and reverse: 5′-AGGAAGGAGGGC TGGAAGAG-3′ for β-actin.
Relative mRNA expression was quantified using the 2−ΔΔCt
method, in which ΔΔCt = (Ctgene−Ctβ)A/B/C-(Ctgene−Ctβ)N. The
relative quantities of the AT1 and AT2
receptors and ACE were normalized to the quantity of the β-actin
transcript in the same sample. All assays were performed in
triplicate.
Statistical analysis
All statistical analyses were performed using SPSS
17.0 (SPSS Inc., Chicago, IL, USA). The incidence of ON was
compared using the χ2 test or Fisher’s exact test. All
data are expressed as the mean ± standard deviation and compared
using one way analysis of variance or the Kruskal-Wallis test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Incidence of steroid-induced ON
Five of the 55 rabbits died following MPA injection
and were excluded from the experiments. One was in group A, two
were in group B and two were in group C. The remaining rabbits were
alive until the end of the experiment. ON was not found in group N,
whereas three of the 14 rabbits in group A, six of the 13 rabbits
in group B and 10 of the 13 rabbits in group C were observed to
develop ON. The incidence of ON in group C (77%) was significantly
higher than that in group A (21%; P=0.007), but not significantly
different from that in group B (46%; P=0.226). Compared with group
A, the incidence of ON in group B was found to increase, but there
was no significant difference between the two groups (P=0.236)
(Fig. 1).
AT1 receptor and ACE protein
expression in bone
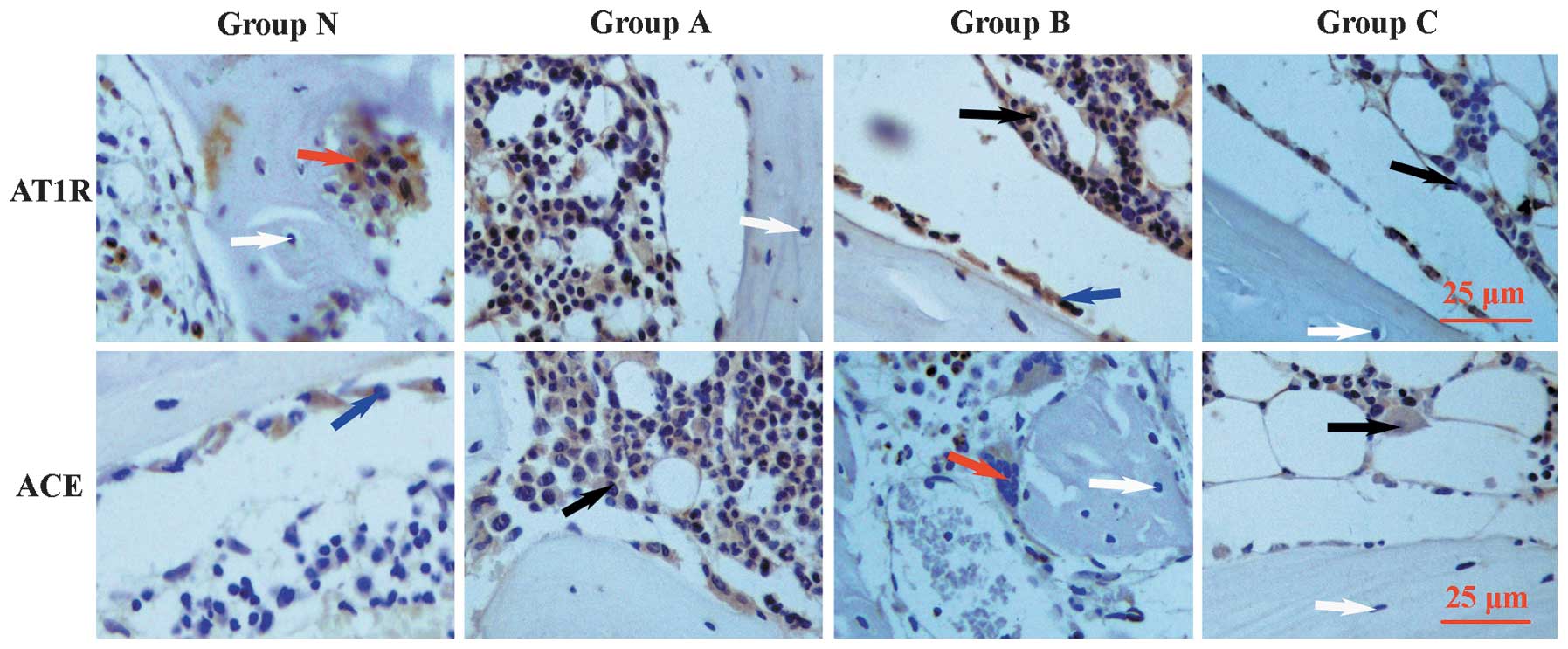
Immunohistological analysis of the femoral heads
showed that the AT1 receptor and ACE were expressed in
the osteoblasts, osteoclasts and bone marrow cells of the bone
tissue, but were not expressed in osteocytes (Fig. 2). This finding is consistent with
previous studies (8,21,22).
Immunostaining for AT1 receptor was observed to increase
in group A and was most significant in group B, compared with group
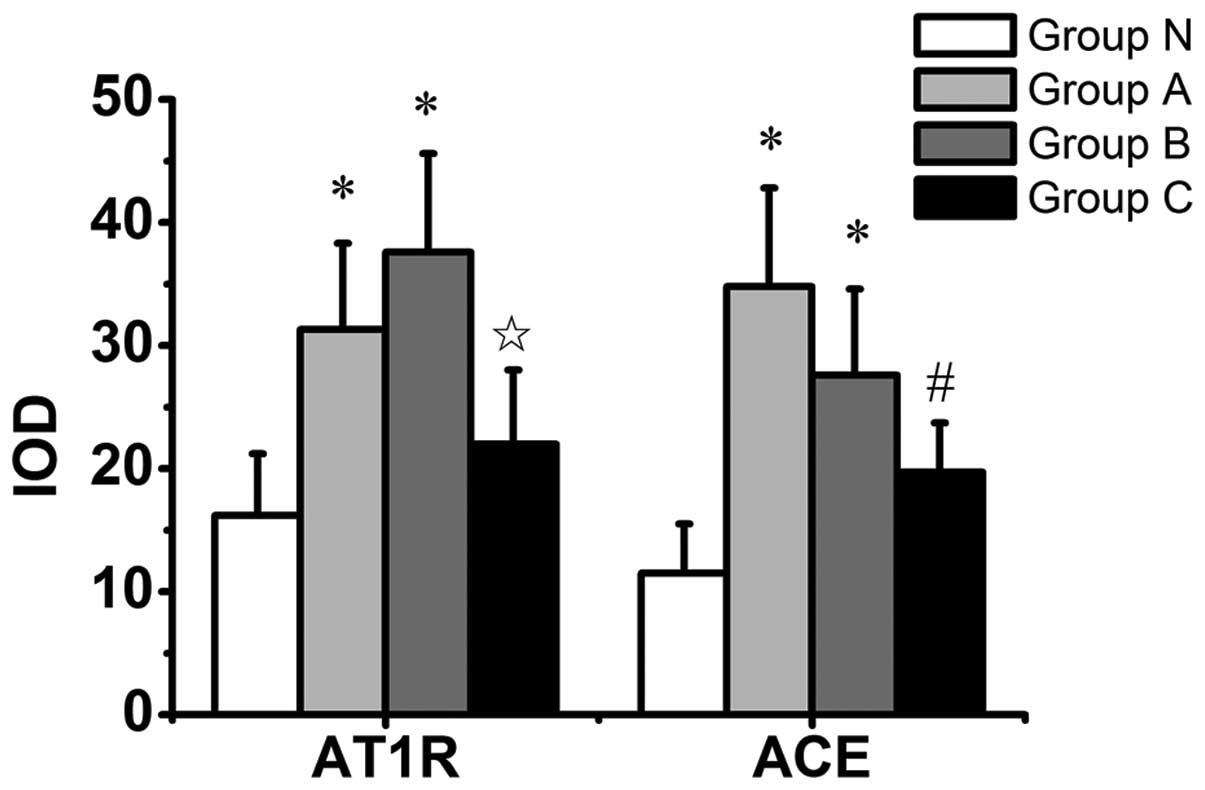
N (Fig. 2). Quantitative image
analysis of AT1 receptor immunostaining revealed an
increase in group A compared with group N (P=0.037), and an
increase in group B compared with groups N and C (P=0.004 and
P=0.032, respectively) (Fig. 3).
Immunostaining for ACE was highest in group A (Fig. 2). Quantitative image analysis of
immunostaining for ACE demonstrated a significant increase in group
A compared with groups N and C (P=0.002 and P=0.037, respectively),
and a significant increase in group B compared with group N
(P=0.026) (Fig. 3).
Western blot analysis revealed a significant
increase in AT1 receptor expression in group A compared
with group N (P=0.004) and in group B compared with groups N and C
(P<0.001). Furthermore, the protein expression of ACE was
observed to increase in group A compared with groups N, B and C
(P<0.001, P=0.023 and P=0.001, respectively), and significantly
increased in groups B and C compared with group N (P=0.002 and
P=0.041, respectively) (Fig.
4).
Ang II concentration and ACE
activity
The concentration of Ang II in the bone was found to
increase in groups A, B and C compared with group N (P=0.001,
P=0.023 and P=0.078, respectively). The concentration of Ang II in
the bone was observed to decline among groups A, B and C; however,
these changes were not significant (Table I). The activity of ACE in the bone
of rabbits in group A was higher than that in groups N, B and C
(P<0.001, P=0.002 and P<0.001, respectively). Furthermore,
the activity of ACE in groups B and C was found to increase
compared with group N (P<0.001). The concentration of Ang II in
the plasma and the activity of ACE in the serum were not observed
to differ significantly among the groups (Table I). These changes in local and
systemic RAS activity are similar to those observed by Mitani et
al (19), who found that
cholesterol increased local tissue ACE activity but not serum ACE
activity in rabbits, suggesting that the systemic RAS may be more
stable than the local RAS.
 | Table IConcentration of Ang II and the
activity of ACE in groups N, A, B and C. |
Table I
Concentration of Ang II and the
activity of ACE in groups N, A, B and C.
| Group | Ang II in bone
(pg/mg pro) | Ang II in plasma
(pg/ml) | ACE activity in
bone (nmol/mg pro/min) | ACE activity in
serum (nmol/ml/min) |
|---|
| N | 1.72±1.09 | 103.81±13.81 | 2.43±0.57 | 139.75±11.33 |
| A | 4.42±2.27a | 99.90±23.11 | 11.92±2.48a | 151.66±16.32 |
| B | 3.48±1.08a | 113.24±17.75 | 8.65±2.46a,b | 131.92±18.31 |
| C | 3.08±1.84a | 102.75±17.34 | 8.07±2.53a,b | 147.70±19.13 |
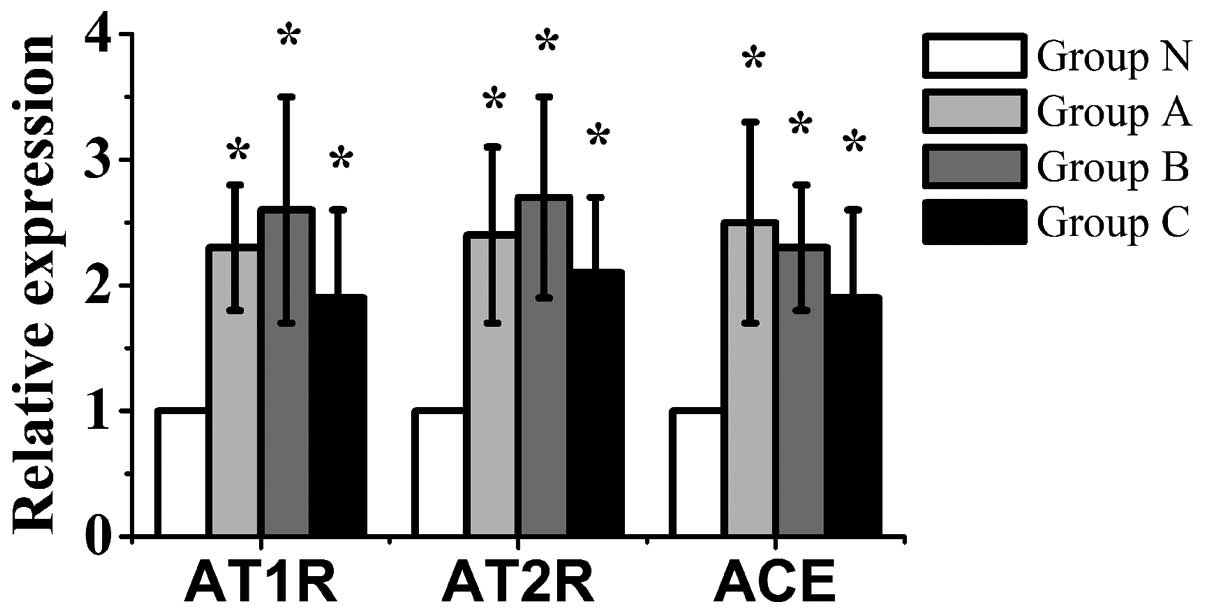
mRNA expression of AT1 and
AT2 receptors and ACE in the bone
The mRNA expression of AT1 and
AT2 receptors and ACE in groups A, B and C was
significantly higher than that in group N (P<0.05). The mRNA
levels of the AT1 and AT2 receptors in group
B were the highest of the 4 groups. Furthermore, the mRNA
expression of ACE in group A was the highest of the 4 groups
(Fig. 5).
Discussion
In the present study, ON was first observed one week
subsequent to MPA injection and was most significant three weeks
following injection. The incidence of ON was 77% three weeks
following MPA administration, which was similar to that observed in
the study by Iwakiri et al (17), in which the incidence of ON was 83%
three weeks subsequent to steroid injection. In the present study,
the expression of Ang II, ACE, and the AT1 and
AT2 receptors significantly increased one week following
MPA administration, concurrent with the onset of ON in this animal
model. Moreover, the expression of Ang II and ACE was highest one
week subsequent to steroid administration and the expression of the
AT1 and AT2 receptors was highest two weeks
following MPA administration. These changes in expression following
MPA injection, precede the time at which ON occurs most
significantly in this animal model, which is three weeks. This
suggests that ON is strongly associated with changes in the
expression of components of the RAS, including Ang II, ACE, and the
AT1 and AT2 receptors.
Although the precise mechanism underlying the
pathogenesis of steroid-induced ON is yet to be elucidated, studies
of the pathophysiology of ON have shown that GCs induce ischemia of
the femoral head and an imbalance in osteoblast and osteoclast
activity, leading to ON, which is highly likely to underlie the
pathogenesis of steroid-induced ON (23–25).
Previous studies have shown that the activation of the local bone
RAS induces metabolic bone disorders (7,9,26)
and impairs bone microcirculation (27). In the present study, the GC MP was
found to activate the local bone RAS, which is similar to the
effect of GCs on the local RAS reported in other tissues (13,16).
Therefore, steroid-induced ON may be closely associated with the
local bone RAS in rabbits.
In the present study, two potential mechanisms were
hypothesized to underlie the activation of the local bone RAS and
induce ON following steroid administration (Fig. 6). The first possible mechanism
involves a disruption in the microcirculation of bone. Ang II is
one of the most potent microvascular vasoactive agents (28), and leads to vessel contraction and
decreased blood supply of the femoral head. Ang II induces the
production of adhesion molecules, chemokines and inflammatory
cytokines, including vascular cell adhesion molecule-1,
intercellular adhesion molecule-1, E-selectin, monocyte
chemoattractant protein-1, interleukin-6, interleukin-8 and tumor
necrosis factor-α in endothelial cells (29). These molecules induce endothelial
cell dysfunction, abnormal blood coagulation and thrombi formation,
which may lead to ischemia of the femoral head (30). Ang II upregulates NADPH oxidase
components, thereby enhancing the production of ROS in the
microcirculation of the bone (31). ROS are potent inter- and
intracellular second messengers, which mediate vessel inflammation
(32). Elevated ROS levels induce
growth arrest and increase the rate of senescence and apoptosis in
endothelial cells. Ang II also stimulates the expression of
plasminogen activator inhibitor-1 (33), which alters homeostatic mechanisms
that balance thrombosis with fibrinolysis and lead to
hypercoagulability of the bone microcirculation.
The activation of the local RAS in the bone
microcirculation may lead to vessel contraction, vascular
inflammation, endothelial cell dysfunction and hypercoagulability
of the bone microcirculation, which decrease the blood supply and
ischemia of the femoral head. These alterations induce a
pathological condition increasing the risk of ON.
The second possible mechanism underlying the
pathogenesis of ON involves altered bone metabolism. In the local
milieu of the bone, two major types of cells, osteoblasts and
osteoclasts, coordinately resorb and form the bone matrix, which
conserves the bone architecture and mass during adulthood (34). However, the balance between
osteoblasts and osteoclasts is disturbed by the activation of the
local bone RAS. Previous studies have shown that Ang II suppresses
osteoblastic cell differentiation and bone formation, and decreases
calcium uptake into the bone (9,10).
In addition, Ang II activates osteoclasts and stimulates bone
resorption (35). In a clinical
study, patients treated with an ACE inhibitor exhibited increased
bone mineral density and reduced fracture risk (11). Therefore, the activation of the
local bone RAS may suppress bone formation and stimulate bone
resorption, causing a bone metabolism disorder through altering the
balance between bone formation and bone resorption, increasing the
risk of ON development and impairing ON repair.
In summary, GCs may activate the local bone RAS,
which may impair bone microcirculation and bone metabolism. These
effects, in conjunction with numerous other factors, including
alterations in lipid metabolism and intraosseous hypertension, may
induce ON.
In the present study, the expression of Ang II, ACE,
and the AT1 and AT2 receptors significantly
increased one week following MPA administration, and the
concentration of Ang II and the activity of ACE in the bone were
highest one week subsequent to MPA injection. However, the
expression of the AT1 and AT2 receptors was
greatest two weeks following MPA injection potentially due to the
overproduction of Ang II one week following MPA injection. Thus,
Ang II may stimulate the expression of the AT1 and
AT2 receptors, which has been reported in a previous
study (36). Therefore, the
administration of ACE inhibitor (ACEI) or angiotensin receptor
blocker (ARB) in rabbits may inhibit the GC-induced activation of
the local bone RAS, which may represent a preventive treatment for
steroid-induced ON. This will be investigated in our future
studies.
The present study has certain limitations. The data
show that the expression of the components of the local bone RAS,
including Ang II, ACE and the AT1 and AT2
receptors were enhanced by MPA in the femoral head, but the precise
role of the local bone RAS in the development of ON requires
further investigation. Thus, future studies will be performed in
order to demonstrate the interrelation between the activation of
local bone RAS and the development of ON, as well as the effect of
ACEI or ARB on preventing steroid-induced ON.
In conclusion, activation of the local bone RAS may
not be the sole cause of steroid-induced ON, and the precise role
of the local bone RAS in the pathogenesis of ON requires further
investigation. However, the present study has contributed to an
enhanced understanding of the molecular processes underlying ON.
Furthermore, these findings suggest the possibility of preventive
approaches to steroid-induced ON through blocking the activation of
the local bone RAS.
Acknowledgements
This study was partially supported by a grant from
the National Natural Science Foundation of China (grant no.
81101337). The authors would like to thank the pathologists Mr.
Xiaoge Zhao and Mr. Gang Cui from the Medical College of Xi’an
Jiaotong University (China) for their contributions.
References
|
1
|
Miyanishi K, Yamamoto T, Irisa T, et al:
Bone marrow fat cell enlargement and a rise in intraosseous
pressure in steroid-treated rabbits with osteonecrosis. Bone.
30:185–190. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ichiseki T, Ueda Y, Katsuda S, Kitamura K,
Kaneuji A and Matsumoto T: Oxidative stress by glutathione
depletion induces osteonecrosis in rats. Rheumatology (Oxford).
45:287–290. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calder JD, Buttery L, Revell PA, Pearse M
and Polak JM: Apoptosis - a significant cause of bone cell death in
osteonecrosis of the femoral head. J Bone Joint Surg Br.
86:1209–1213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ichiseki T, Matsumoto T, Nishino M,
Kaneuji A and Katsuda S: Oxidative stress and vascular permeability
in steroid-induced osteonecrosis model. J Orthop Sci. 9:509–515.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kabata T, Kubo T, Matsumoto T, et al:
Onset of steroid-induced osteonecrosis in rabbits and its
relationship to hyperlipaemia and increased free fatty acids.
Rheumatology (Oxford). 44:1233–1237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paul M, Poyan Mehr AP and Kreutz R:
Physiology of local renin-angiotensin systems. Physiol Rev.
86:747–803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asaba Y, Ito M, Fumoto T, et al:
Activation of renin-angiotensin system induces osteoporosis
independently of hypertension. J Bone Miner Res. 24:241–250. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garcia P, Schwenzer S, Slotta JE, et al:
Inhibition of angiotensin-converting enzyme stimulates fracture
healing and periosteal callus formation - role of a local
renin-angiotensin system. Br J Pharmacol. 159:1672–1680. 2010.
View Article : Google Scholar
|
|
9
|
Schurman SJ, Bergstrom WH, Shoemaker LR
and Welch TR: Angiotensin II reduces calcium uptake into bone.
Pediatr Nephrol. 19:33–35. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hagiwara H, Hiruma Y, Inoue A, Yamaguchi A
and Hirose S: Deceleration by angiotensin II of the differentiation
and bone formation of rat calvarial osteoblastic cells. J
Endocrinol. 156:543–550. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lynn H, Kwok T, Wong SY, Woo J and Leung
PC: Angiotensin converting enzyme inhibitor use is associated with
higher bone mineral density in elderly Chinese. Bone. 38:584–588.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marchesi C, Paradis P and Schiffrin EL:
Role of the renin-angiotensin system in vascular inflammation.
Trends Pharmacol Sci. 29:367–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fishel RS, Eisenberg S, Shai SY, Redden
RA, Bernstein KE and Berk BC: Glucocorticoids induce
angiotensin-converting enzyme expression in vascular smooth muscle.
Hypertension. 25:343–349. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barreto-Chaves M, Anéas I and Krieger J:
Glucocorticoid regulation of angiotensin-converting enzyme in
primary culture of adult cardiac fibroblasts. Am J Physiol Regul
Integr Comp Physiol. 280:R25–R32. 2001.PubMed/NCBI
|
|
15
|
Mendelsohn FA, Lloyd CJ, Kachel C and
Funder JW: Induction by glucocorticoids of angiotensin-converting
enzyme production from bovine endothelial cells in culture and rat
lung in vivo. J Clin Invest. 70:684–692. 1982. View Article : Google Scholar
|
|
16
|
Sato A, Suzuki H, Nakazato Y, Shibata H,
Inagami T and Saruta T: Increased expression of vascular
angiotensin II type 1A receptor gene in glucocorticoid-induced
hypertension. J Hypertens. 12:511–516. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iwakiri K, Oda Y, Kaneshiro Y, et al:
Effect of simvastatin on steroid-induced osteonecrosis evidenced by
the serum lipid level and hepatic cytochrome P4503A in a rabbit
model. J Orthop Sci. 13:463–468. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuribayashi M, Fujioka M, Takahashi KA, et
al: Vitamin E prevents steroid-induced osteonecrosis in rabbits.
Acta Orthop. 81:154–160. 2010. View Article : Google Scholar
|
|
19
|
Mitani H, Bandoh T, Ishikawa J, Kimura M,
Totsuka T and Hayashi S: Inhibitory effects of fluvastatin, a new
HMG-CoA reductase inhibitor, on the increase in vascular ACE
activity in cholesterol-fed rabbits. Br J Pharmacol. 119:1269–1275.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu BC, Gao J, Li Q and Xu LM: Albumin
caused the increasing production of angiotensin II due to the
dysregulation of ACE/ACE2 expression in HK2 cells. Clin Chim Acta.
403:23–30. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Izu Y, Mizoguchi F, Kawamata A, et al:
Angiotensin II type 2 receptor blockade increases bone mass. J Biol
Chem. 284:4857–4864. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haznedaroglu IC and Oztürk MA: Towards the
understanding of the local hematopoietic bone marrow
renin-angiotensin system. Int J Biochem Cell Biol. 35:867–880.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Powell C, Chang C and Gershwin ME: Current
concepts on the pathogenesis and natural history of steroid-induced
osteonecrosis. Clin Rev Allergy Immunol. 41:102–113. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Wang N, Li M and Wang KZ: To
investigate the role of the nervous system of bone in
steroid-induced osteonecrosis in rabbits. Osteoporos Int.
21:2057–2066. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drescher W, Weigert KP, Bunger MH,
Ingerslev J, Bünger C and Hansen ES: Femoral head blood flow
reduction and hypercoagulability under 24 h megadose steroid
treatment in pigs. J Orthop Res. 22:501–508. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sabanai K, Tsutsui M, Sakai A, et al:
Genetic disruption of all NO synthase isoforms enhances BMD and
bone turnover in mice in vivo: involvement of the renin-angiotensin
system. J Bone Miner Res. 23:633–643. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fukuda D and Sata M: Role of bone marrow
renin-angiotensin system in the pathogenesis of atherosclerosis.
Pharmacol Ther. 118:268–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wassmann S and Nickenig G:
Pathophysiological regulation of the AT1-receptor and implications
for vascular disease. J Hypertens Suppl. 24:S15–S21. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pueyo ME, Gonzalez W, Nicoletti A, Savoie
F, Arnal JF and Michel JB: Angiotensin II stimulates endothelial
vascular cell adhesion molecule-1 via nuclear factor-kappaB
activation induced by intracellular oxidative stress. Arterioscler
Thromb Vasc Biol. 20:645–651. 2000. View Article : Google Scholar
|
|
30
|
Harrison DG: Cellular and molecular
mechanisms of endothelial cell dysfunction. J Clin Invest.
100:2153–2157. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yao EH, Fukuda N, Matsumoto T, et al:
Losartan improves the impaired function of endothelial progenitor
cells in hypertension via an antioxidant effect. Hypertens Res.
30:1119–1128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ohtsu H, Frank GD, Utsunomiya H and Eguchi
S: Redox-dependent protein kinase regulation by angiotensin II:
mechanistic insights and its pathophysiology. Antioxid Redox
Signal. 7:1315–1326. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vaughan DE, Lazos SA and Tong K:
Angiotensin II regulates the expression of plasminogen activator
inhibitor-1 in cultured endothelial cells. A potential link between
the renin-angiotensin system and thrombosis. J Clin Invest.
95:995–1001. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao C, Irie N, Takada Y, et al:
Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis.
Cell Metab. 4:111–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shimizu H, Nakagami H, Osako MK, et al:
Angiotensin II accelerates osteoporosis by activating osteoclasts.
FASEB J. 22:2465–2475. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu JJ, Li DL, Zhou J, et al:
Acetylcholine prevents angiotensin II-induced oxidative stress and
apoptosis in H9c2 cells. Apoptosis. 16:94–103. 2011. View Article : Google Scholar : PubMed/NCBI
|