Introduction
Apelin is a novel adipocyte-derived factor that
binds the orphan G protein-coupled receptor (GPCR) APJ with high
affinity (1). Apelin mRNA encodes
a 77-amino-acid prepropeptide, which is proteolytically cleaved to
yield bioactive peptides of 36, 17 and 13 amino acids (aa) in size.
Each of these peptides contains the extreme C-terminal region of
the precursor protein and their bioactivity resides in the
C-terminal 13 aa fragment (2). The
sequence of the 13 aa apelin peptide is highly conserved among
different species, suggesting that its critical function has been
evolutionarily conserved (3).
The apelinergic system has a widespread pattern of
distribution (4,5). However, the majority of studies
regarding the function of apelin/APJ are on the cardiovascular
system, due to its similarity to the angiotensin receptor. In the
cardiovascular system, apelin reduces blood pressure and modulates
the contractility of cardiac tissue and blood vessels (6,7,8).
Apelin signaling is also associated with tumor angiogenesis and
vascular regeneration during embryogenesis (9).
AMP-activated protein kinase (AMPK) is a
stress-activated protein kinase, which is involved in the
regulation of energy and metabolic homeostasis (10). In vascular endothelium, AMPK
signaling is required for vascular endothelial growth factor
(VEGF)-induced nitric oxide (NO) production, migration and
differentiation under conditions of hypoxia (11). Previous studies indicated that
apelin-13 is able to regulate glucose and lipid metabolism
(12,13) via the activation of AMPK signaling.
The disruption of apelin-13 signaling can increase the
hypoxia-induced pulmonary hypertension mediated by decreased
activation of AMPK and endothelial nitric oxide synthase (eNOS)
(14). In addition, as protein
kinase Akt/protein kinase B (Akt) signaling participates in
vascular homeostasis and angiogenesis (15,16),
it can also phosphorylate eNOS (17), resulting in NO production and
subsequently leading to the regulation of vasomotor responses.
Thus, it was hypothesized that AMPK and Akt signaling may
participate in the regulation of angiogenesis stimulated by
apelin-13.
The aim of the present study was to investigate the
role of apelin-13 in angiogenesis in rat primary myocardial
microvascular endothelial cells (MMVECs) and the mechanisms
involved in it. In addition, compound C, an AMPK inhibitor, and
LY294002, an Akt inhibitor were used to investigate whether AMPK
and Akt signaling participate in the pro-angiogenic process induced
by apelin-13.
Materials and methods
Materials
Apelin-13 was purchased from GL Biochem Shanghai
(Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM) and
fetal bovine serum (FBS) were purchased from Gibco-BRL (Carlsbad,
CA, USA). Phospho-AMPK (Thr-172), pan-α-AMPK, phospho-Akt (Ser-473)
and phospho-eNOS (Ser-1179) antibodies were purchased from Cell
Signaling Technology (Beverly, MA, USA). Akt, eNOS and GAPDH
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Compound C was purchased from Toronto
Research Chemicals Inc. (North York, ON, Canada) and LY294002 from
Calbiochem (San Diego, CA, USA). Secondary antibodies were
purchased from Beijing Biosynthesis Biotechnology Co., Ltd.
(Beijing, China). All antibodies were polyclonal, and sourced from
rabbit.
Isolation and identification of MMVECs in
rats
Wistar rats (80–100 g) were used for the isolation
of primary MMVECs. The animal protocol was approved by the Animal
Care Committee of Shanghai Jiao Tong University (Shanghai, China).
Briefly, rats were anesthetized with sodium pentobarbital (60
mg/kg) and heparinized by intraperitoneal injection of sodium
heparin (500 U/0.1 kg). Following thoracotomy, the heart was
rapidly dislodged and washed in phosphate-buffered saline (PBS).
The atria, visible connective tissue, the right ventricle and the
epicardial and endocardial surfaces of the left ventricle were
carefully removed and the remaining myocardial tissue was washed in
PBS with antibiotics (penicillin-streptomycin; 15140-122; Life
Technologies, Carlsbad, CA, USA) several times prior to cutting
into 1-mm3 sections without visible vessels. Myocardial
tissues were seeded on culture plates pre-coated with rat tail
tendon gelatin and incubated at 37°C in a humidified atmosphere of
5% CO2 for 30 min. The tissues were cultured in DMEM
(4500 mg/l; D-glucose) supplemented with 20% FBS, 50 U/ml heparin,
100 U/ml penicillin and 100 μg/ml streptomycin. The tissue sections
were discarded after the cells began to grow and the medium was
changed at 70 h intervals. MMVECs were identified by their typical
‘cobblestone’ appearance and by positive CD31 and CD34
immunostaining. MMVECs at the second passage were used for
experiments. The cells were allowed to grow to 80–90% confluence
and were used for further experimental analysis.
Cell proliferation assay
The proliferation rate of MMVECs was determined by
the MTT assay. The cells were trypsinized and resuspended in 10%
DMEM. The cells were seeded at 2×103 cells per well in
96-well plates and cultured in a humidified 5% CO2
atmosphere at 37°C for 24 h. Drugs (apelin-13, compound C and
LY294002) were added to the medium at the indicated times with
different concentrations. Following incubation, the cells were
washed with PBS and subsequently incubated with MTT at 37°C for 4
h. Following incubation, dimethylsulfoxide was added, the cells
were incubated for 10 min and the absorbance at 490 nm was recorded
with a Epoch Microplate spectrophotometer (BioTek Instruments,
Inc., Winooski, VT, USA).
Migration assays
Cell scratch assay
MMVECs were trypsinized and resuspended in 10% DMEM.
The cells were incubated in 6-well culture dishes in DMEM
containing 10% FBS until 90% confluency. Following serum starvation
for 24 h, a linear wound was made by scratching the bottom of the
dish. Following wounding, cells were washed with PBS and incubated
in serum-free DMEM. Following treatment, cells were allowed to
migrate for 0, 8, 16 and 24 h. The wound width was measured in
three areas selected at random and images were captured using the
Motic AE31 Photometry and Dimensioning microscope (Milton, MA,
USA). Migration was quantified by the assessment of the migration
distance beyond the reference line using the Motic Image Plus
software (Houston, TX, USA).
Boyden chamber assay
The Boyden chamber assay was performed as described
previously (Neuroprobe, Cabin John, MD, USA) (18). Cells were trypsinized and
resuspended in 10% DMEM. DMEM (800 μl/well; 10%) containing various
concentrations of apelin-13, compound C and LY294002 was added into
the wells in the lower chamber and 1.5×104 cells (200
μl/well) were added in the upper chamber. The chambers were
incubated for 8–12 h at 37°C in a 5% CO2 humidified
incubator. The cells migrating through the filter (cells on the
lower side of the filter) were fixed in 4% paraformaldehyde for 10
min, stained with 0.1% crystal violet stain solution (Sigma, St.
Louis, MO, USA) for 30 min and then five random microscopic fields
per well were quantified. Each experiment was performed twice in
triplicate.
Tube formation assay
The formation of vascular-like structures by MMVECs
on growth factor-reduced Matrigel (BD Biosciences, Franklin Lakes,
NJ, USA) was performed as previously described (19) and 24-well culture plates were
coated with Matrigel according to the manufacturer’s instructions.
The MMVECs were seeded on coated plates at 5×104
cells/well in DMEM containing 10% FBS and incubated at 37°C for
16–18 h. Tube formation was observed using an inverted phase
contrast microscope (Nikon, Tokyo, Japan). Images were captured
with a video graphic system (DEI-750 CE Digital Output Camera;
Optronics, Goleta, CA, USA). The degree of tube formation was
quantified by measuring the length of tubes from three randomly
selected low power fields (x100) from each well using the NIH Image
Program. Each experiment was repeated three times.
Western blot analysis
Western blot analysis was performed as previously
described (20). In brief, cell
lysates were extracted with Nonidet P-40 lysis buffer followed by
SDS-PAGE. The membranes were immunoblotted with the indicated
primary antibodies at a 1:1,000 dilution overnight, followed by the
secondary antibody conjugated with horseradish peroxidase at a
1:2,000 dilution for 2 h. A western blotting detection kit
(Amersham Biosciences, Piscataway, NJ, USA) and enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Waltham, MA,
USA) were used to examine the membranes. Protein bands were
evaluated by densitometry using ImageJ software (version 1.41;
National Institutes of Health, Bethesda, MD, USA) and normalized to
the expression levels of GAPDH for protein loading.
Phospho-specific signals, normalized against the quantity of total
protein, are shown as arbitrary units. The changes in
phosphorylation were calculated by comparing the differences
between basal and stimulated values of specifically treated samples
and their respective controls.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Statistical comparisons were performed using analysis of
variance with Scheffe’s F-test for post hoc analysis. P<0.05 was
considered to indicate a statistically significant difference.
Statistical analyses were performed using the SPSS 13.0 statistical
software (SPSS, Chicago, IL, USA).
Results
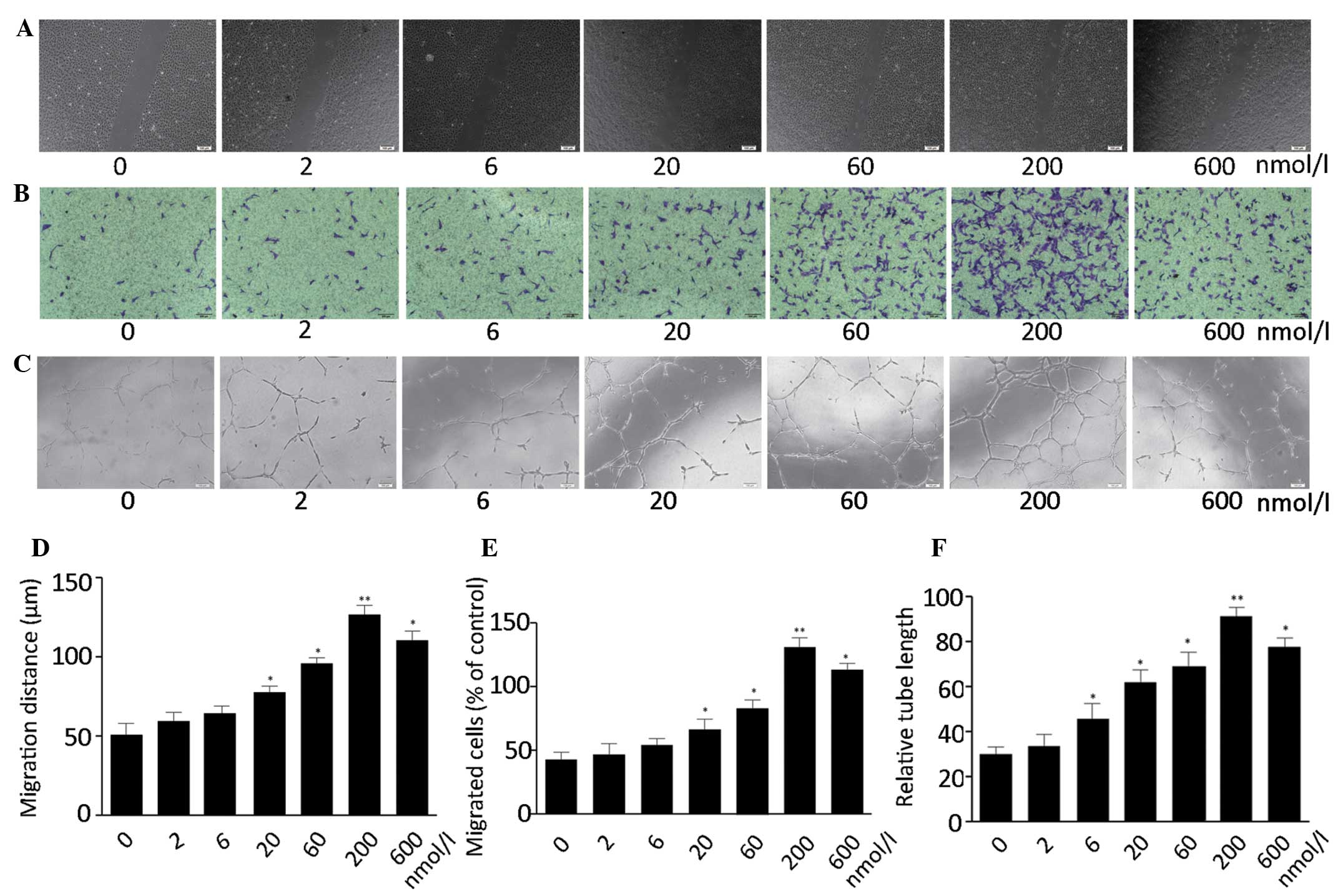
Apelin-13 promotes the proliferation,
migration and tube formation of MMVECs
The effect of apelin-13 on the proliferation of
MMVECs was examined using the MTT assay. It was revealed that
apelin-13 was able to promote the proliferation of MMVECs in a
dose-dependent manner with a maximum effect being observed at 200
nmol/l (Fig. 1). The role of
apelin-13 on endothelial cell migration was further evaluated by
the scratch assay and the modified Boyden chamber assay. Apelin-13
significantly stimulated the migration of cells in a dose-dependent
manner with an optimal activity at 200 nmol/l (Fig. 2A and B). To examine the effects of
apelin-13 on the differentiation of MMVECs into vascular structures
in vitro, tube formation ability was assessed. Apelin-13
significantly increased the tube formation of MMVECs in a
dose-dependent manner with its optimal activity at 200 nmol/l
(Fig. 2C).
Apelin-13 induces the phosphorylation of
AMPK and Akt in MMVECs
It is known that AMPK and Akt are involved in the
regulation of angiogenesis (11,15).
The effect of apelin-13 on the phosphorylation status of AMPK at
Thr-172 and Akt at Ser-473 was investigated in MMVECs. The
treatment of MMVECs with apelin-13 enhanced the phosphorylation of
AMPK in a dose- and time-dependent manner with maximal AMPK
phosphorylation occurring at 200 nmol/l following 2 h exposure.
Similarly, the phosphorylation of Akt was induced by apelin-13;
however, this response was transient (Fig. 3A and C). Cross-talk between AMPK
and Akt was previously described to regulate the phosphorylation of
eNOS (11,21). Therefore, eNOS phosphorylation in
MMVECs was also examined. Apelin-13 stimulated eNOS phosphorylation
at Ser-1179 (p-eNOS) in a time- and dose-dependent manner, while
the total protein levels (eNOS) remained unchanged. (Fig. 3A and C). Subsequently, MMVECs were
treated with the AMPK and Akt inhibitors, it was revealed that they
significantly suppressed the apelin-13-induced AMPK, Akt and eNOS
phosphorylation in MMVECs (Fig. 3E and
G), while incubation with the Akt inhibitor did not alter the
endogenous phosphorylation of AMPK (Fig. 3G). Therefore, it was hypothesized
that there is cross-talk between AMPK and Akt in the pro-angiogenic
process induced by apelin-13.
AMPK and Akt signaling are required for
apelin-13-stimulated migration and differentiation
To determine whether AMPK and Akt signaling are
involved in apelin-13-stimulated endothelial cell migration and
tube formation in vitro, MMVECs were treated either with
compound C or LY294002, the inhibitors of AMPK and Akt,
respectively. Cell scratch and Boyden chamber assays were used to
assess the role of AMPK and Akt signaling on the migration ability
of MMVECs induced by apelin-13. It was observed that the AMPK and
Akt inhibitors significantly suppressed the apelin-13-stimulated
migration of MMVECs (Fig. 4A and
C). Subsequently, Matrigel assays were performed to investigate
the effect of these two inhibitors on the tube formation induced by
apelin-13 in MMVECs. The inhibitors significantly impaired the
apelin-13-stimulated tube formation (Fig. 4E) of MMVECs. These data indicated
that AMPK and Akt signaling are required for apelin-13-induced
endothelial migration and differentiation in MMVECs.
Discussion
In the present study, it was demonstrated that
apelin-13 promotes angiogenesis through the modulation of AMPK and
Akt signaling in MMVECs. It was further investigated how AMPK and
Akt signaling participates in the pro-angiogenic process and it was
found that the activation of eNOS is involved in the two
processes.
The regulatory role of apelin-13 in endothelial
cells has previously been investigated. It was revealed that apelin
is expressed in vascular endothelial cells and participates in
regulating their proliferation and regenerative angiogenesis
(22,23,24).
Calcitonin gene-related peptide and vascular endothelial growth
factor were reported to promote angiogenesis by promoting AMPK and
Akt signaling activation, respectively (25,26).
AMPK and Akt directly phosphorylate eNOS (17,27),
which is important in angiogenesis. By contrast, apelin-13 can
regulate glucose and lipid metabolism (12,13)
via activating AMPK signaling. We hypothesized that apelin-13 may
promote angiogenesis through the activation of AMPK and Akt
signaling in MMVECs. The results demonstrated that apelin-13 can
promote angiogenesis in MMVECs. Apelin-13 stimulated the
phosphorylation of AMPK and eNOS in a time- and dose-dependent
manner, while the phosphorylation of Akt increased in the early
stages and decreased at the later stages. AMPK and Akt inhibitors
suppressed the apelin-13-induced AMPK, Akt and eNOS phosphorylation
and also inhibited apelin-13-stimulated cell migration and
differentiation. Notably, although the AMPK inhibitor suppressed
the endogenous Akt phosphorylation, the Akt inhibitor had no effect
on the endogenous phosphorylation of AMPK in MMVECs. The present
data suggest that apelin-13 exerts pro-angiogenic effects in MMVECs
through the modulation of AMPK and Akt signaling and the activation
of eNOS, which is in accordance with previous studies (28,29,30).
A study by Nagata et al (11) demonstrated that AMPK did not
exhibit an effect on endothelial cell migration, tube formation and
NO production under normoxia, which suggests that further in
vitro and in vivo studies are required to gain a greater
understanding of the role of endothelial cell AMPK in
angiogenesis.
Of note, in the present study, it was not determined
how apelin-13 regulates AMPK and Akt signaling. In addition,
although a subunit of AMPK was identified as the major target,
further evaluation is required regarding the specific subunit (α1
or α2) involved. However, it was demonstrated that the activation
of AMPK and Akt signaling was associated with angiogenesis induced
by apelin-13 in MMVECs. Notably, the impaired angiogenesis was
associated with the inhibition of the pathways.
The imbalance between angiogenesis and cardiac
hypertrophy is important in the transition from adaptive
hypertrophy to pathological cardiac remodeling in heart failure.
Apelin-13 has been demonstrated to exert cardiovascular protective
effects (31,32). The results of the present study
demonstrated that apelin-13 is a pro-angiogenic factor, suggesting
that the exogenous supplementation of apelin-13 may improve cardiac
remodeling in patients with heart failure.
Acknowledgements
This study was supported by the Natural Science
Foundation of Shanghai Science and Technology Commission (grant no.
10ZR1422900 to Professor Mingya Liu) and partly supported by the
National Natural Science Foundation of China (grant no. 81070110 to
Professor Meng Wei).
References
|
1
|
Tatemoto K, Hosoya M, Habata Y, et al:
Isolation and characterization of a novel endogenous peptide ligand
for the human APJ receptor. Biochem Biophys Res Commun.
251:471–476. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kawamata Y, Habata Y, Fukusumi S, et al:
Molecular properties of apelin: tissue distribution and receptor
binding. Biochim Biophys Acta. 1538:162–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kälin RE, Kretz MP, Meyer AM, et al:
Paracrine and autocrine mechanisms of apelin signaling govern
embryonic and tumor angiogenesis. Dev Biol. 305:599–614.
2007.PubMed/NCBI
|
|
4
|
Hosoya M, Kawamata Y, Fukusumi S, et al:
Molecular and functional characteristics of APJ. Tissue
distribution of mRNA and interaction with the endogenous ligand
apelin. J Biol Chem. 275:21061–21067. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O’Carroll AM, Selby TL, Palkovits M and
Lolait SJ: Distribution of mRNA encoding B78/apj, the rat homologue
of the human APJ receptor, and its endogenous ligand apelin in
brain and peripheral tissues. Biochim Biophys Acta. 1492:72–80.
2000.PubMed/NCBI
|
|
6
|
Cheng X, Cheng XS and Pang CC: Venous
dilator effect of apelin, an endogenous peptide ligand for the
orphan APJ receptor, in conscious rats. Eur J Pharmacol.
470:171–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Szokodi I, Tavi P, Földes G, et al:
Apelin, the novel endogenous ligand of the orphan receptor APJ,
regulates cardiac contractility. Circ Res. 91:434–440. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Farkasfalvi K, Stagg MA, Coppen SR, et al:
Direct effects of apelin on cardiomyocyte contractility and
electrophysiology. Biochem Biophys Res Commun. 357:889–895. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ribatti D, Vacca A, Roncali L and Dammacco
F: The chick embryo chorioallantoic membrane as a model for in vivo
research on angiogenesis. Int J Dev Biol. 40:1189–1197.
1996.PubMed/NCBI
|
|
10
|
Mu J, Brozinick JT Jr, Valladares O, Bucan
M and Birnbaum MJ: A role for AMP-activated protein kinase in
contraction- and hypoxia-regulated glucose transport in skeletal
muscle. Mol Cell. 7:1085–1094. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nagata D, Mogi M and Walsh K:
AMP-activated protein kinase (AMPK) signaling in endothelial cells
is essential for angiogenesis in response to hypoxic stress. J Biol
Chem. 278:31000–31006. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu S, Sun F, Li W, et al: Apelin
stimulates glucose uptake through the PI3K/Akt pathway and improves
insulin resistance in 3T3-L1 adipocytes. Mol Cell Biochem.
353:305–313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yue P, Jin H, Xu S, et al: Apelin
decreases lipolysis via G(q), G(i), and AMPK-Dependent Mechanisms.
Endocrinology. 152:59–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chandra SM, Razavi H, Kim J, et al:
Disruption of the apelin-APJ system worsens hypoxia-induced
pulmonary hypertension. Arterioscler Thromb Vasc Biol. 31:814–820.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shiojima I and Walsh K: Role of Akt
signaling in vascular homeostasis and angiogenesis. Circ Res.
90:1243–1250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morales-Ruiz M, Fulton D, Sowa G, Languino
LR, Fujio Y, Walsh K and Sessa WC: Vascular endothelial growth
factor-stimulated actin reorganization and migration of endothelial
cells is regulated via the serine/threonine kinase Akt. Circ Res.
86:892–896. 2000. View Article : Google Scholar
|
|
17
|
Luo Z, Fujio Y, Kureishi Y, et al: Acute
modulation of endothelial Akt/PKB activity alters nitric
oxide-dependent vasomotor activity in vivo. J Clin Invest.
106:493–499. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Witzenbichler B, Kureishi Y, Luo Z, Le
Roux A, Branellec D and Walsh K: Regulation of smooth muscle cell
migration and integrin expression by the Gax transcription factor.
J Clin Invest. 104:1469–1480. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kureishi Y, Luo Z, Shiojima I, et al: The
HMG-CoA reductase inhibitor simvastatin activates the protein
kinase Akt and promotes angiogenesis in normocholesterolemic
animals. Nat Med. 6:1004–1010. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagata D, Suzuki E, Nishimatsu H, et al:
Cyclin A downregulation and p21(cip1) upregulation correlate with
GATA-6-induced growth arrest in glomerular mesangial cells. Circ
Res. 87:699–704. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kovacic S, Soltys CL, Barr AJ, Shiojima I,
Walsh K and Dyck JR: Akt activity negatively regulates
phosphorylation of AMP-activated protein kinase in the heart. J
Biol Chem. 278:39422–39427. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kleinz MJ and Davenport AP:
Immunocytochemical localization of the endogenous vasoactive
peptide apelin to human vascular and endocardial endothelial cells.
Regul Pept. 118:119–125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eyries M, Siegfried G, Ciumas M, Montagne
K, Agrapart M, Lebrin F and Soubrier F: Hypoxia-induced apelin
expression regulates endothelial cell proliferation and
regenerative angiogenesis. Circ Res. 103:432–440. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Masri B, Morin N, Cornu M, Knibiehler B
and Audigier Y: Apelin (65–77) activates p70 S6 kinase and is
mitogenic for umbilical endothelial cells. FASEB J. 18:1909–1911.
2004.
|
|
25
|
Zheng S, Li W, Xu M, et al: Calcitonin
gene-related peptide promotes angiogenesis via AMP-activated
protein kinase. Am J Physiol Cell Physiol. 299:C1485–C1492. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Radisavljevic Z, Avraham H and Avraham S:
Vascular endothelial growth factor up-regulates ICAM-1 expression
via the phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway
and modulates migration of brain microvascular endothelial cells. J
Biol Chem. 275:20770–20774. 2000. View Article : Google Scholar
|
|
27
|
Morrow VA, Foufelle F, Connell JM, Petrie
JR, Gould GW and Salt IP: Direct activation of AMP-activated
protein kinase stimulates nitric-oxide synthesis in human aortic
endothelial cells. J Biol Chem. 278:31629–31639. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu Z, Fu C, Li X, et al: Prostaglandin E2
promotes endothelial differentiation from bone marrow-derived cells
through AMPK activation. PLoS One. 6:e235542011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su KH, Yu YB, Hou HH, et al: AMP-activated
protein kinase mediates erythropoietin-induced activation of
endothelial nitric oxide synthase. J Cell Physiol. 227:3053–3062.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Izumi Y, Shiota M, Kusakabe H, et al:
Pravastatin accelerates ischemia-induced angiogenesis through
AMP-activated protein kinase. Hypertens Res. 32:675–679. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tao J, Zhu W, Li Y, et al: Apelin-13
protects the heart against ischemia-reperfusion injury through
inhibition of ER-dependent apoptotic pathways in a time-dependent
fashion. Am J Physiol Heart Circ Physiol. 301:H1471–H1486. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rastaldo R, Cappello S, Folino A, et al:
Apelin-13 limits infarct size and improves cardiac postischemic
mechanical recovery only if given after ischemia. Am J Physiol
Heart Circ Physiol. 300:H2308–H2315. 2011. View Article : Google Scholar : PubMed/NCBI
|