Introduction
Down’s syndrome (DS) or trisomy 21 is a chromosomal
condition caused by the presence of all or part of an extra 21st
chromosome. DS is a high-incidence birth defect that is often
associated with impairment of cognitive ability and physical growth
(1). Individuals with DS have a
higher risk of congenital heart disease (CHD), dysfunction of the
thyroid gland, Hirschsprung’s disease, eye and hearing disorders,
leukemia and testicular cancer; however, there is a wide range of
phenotypic variation in DS (2–6). It
is now more than half a century since DS was first shown to result
from trisomy 21 (7). Although
progress has been made by investigating genes to understand the
complex phenotypes associated, the mechanisms remain far from
clear.
DS disorders are the result of extra copies of the
genes located on chromosome 21. In general, an overexpression of
the genes arises and DNA hypomethylation is a possible mechanism to
explain the altered gene expression. DNA methylation often occurs
in a CpG dinucleotide, in which the cytosine gains a methyl group.
Hypermethylation results in transcriptional silencing, for example
genomic imprinting and X-chromosome inactivation, while
hypomethylation is linked to chromosomal instability and loss of
imprinting. Unmethylated CpGs are often grouped in CpG islands
(CGIs), which are present in the 5′ regulatory regions of a number
of genes and acquire abnormal hypermethylation in numerous disease
processes. Alterations of DNA methylation have been recognized as
an important component of cancer development (8). Altered methylation status in
peripheral blood lymphocytes (PBLs) has been linked to increased
risk of several diseases and, in addition, PBLs are an easily
accessible source to identify potential epigenetic biomarkers
(9).
In the present study, a comprehensive CGI
methylation analysis was performed using trisomy 21 and control
cases to identify the allelic methylation status in CGIs of PBLs.
The aim of the study was to determine the utility of CGI
methylation analysis associated with human diseases, in particular,
the DS complex phenotypes and to locate potential epigenetic
biomarkers for prenatal diagnosis. To the best of our knowledge,
there are no comprehensive CGI methylation analyses that have
focused on chromosome 21q using a large number of trisomy 21
samples (10), thus, the present
study focused on chromosome 21, which is the major contributor of
DS complex phenotype.
Materials and methods
Alignment of CGI data to the BGI YH
database
The present analysis was based on the results of a
comprehensive measurement of CGI methylation on human chromosome
21q (11). Yamada et al
repeat-masked the chromosome sequence and computationally
identified all non-repetitive CGIs using standard tools and
parameters (GC content, >50%; ratio of observed versus expected
number of CpG dinucleotides, >0.6; >400 base pairs in
length). The authors designed primers for the 149 CGI identified
and extracted corresponding DNA from samples of human PBLs.
Finally, the authors determined the methylation status of each CGI
using methylation-specific restriction enzymes (via
HpaII-McrBC-PCR). The DNA sequences of 149 CGIs were
acquired from UCSC (11) and were
aligned with the sequence in BLAST, the BGI YH database (http://yh.genomics.org.cn/search.jsp).
The score of each alignment was indicated by one of five colors, in
which the highest score was >200 and shown in red. Multiple
alignments on the same database sequence were connected by a
striped line. A continuous red line indicated a perfect match (100%
match) and the alignment data was classified into ten groups based
upon the percentage of red line. Subsequently, the primers of the
148 CGIs (11) were aligned in
sequence in the BLAST BGI YH database (CGI no. 103 was investigated
with bisulfite sequencing as it lacked HpaII and HhaI
sites and could not detected by HM-PCR). When a base differed to a
sequence in the database, the primer was a 0% match. The alignment
results were divided into three groups: Perfect match (a pair of
primers was 100% matched); 50% match (one primer of a pair was 100%
matched); and no match (a pair of primers was 0% matched).
Study subjects and diagnosis
A total of 150 control cases and 150 DS cases were
obtained through the Children’s Hospital of Chongqing Medical
University (Chongqing, China) and participant’s families or
correspondents provided informed consent. The distribution of age
and residential placement did not differ between the control and
the patients. Confirmation of trisomy 21 was obtained by G-banded
karyotypes, all patients had complete trisomy 21, with 100%
concordance between cytogenetics and the clinical diagnosis of DS.
Data for Japanese individuals were obtained from the study by
Yamada et al (11).
Preparation of genomic DNA from human
PBLs
Trisomy 21 and normal human lymphocytes were
prepared from peripheral blood. Total genomic DNA was extracted by
TIANamp Blood DNA kit (Tiangen Biotech, Beijing, China) following
the manufacturer’s instructions. The concentration of DNA was
determined using the GeneQuant pro RNA/DNA Calculator (GE
Healthcare, Pittsburgh, PA, USA) and the integrity of DNA was
determined by electrophoresis.
HpaII-McrBC PCR assay
Human genomic DNA (0.5 μg) was digested with 15
units HpaII or HhaI (Promega Corp., Madison, WI, USA)
or 100 units McrBC (New England Biolabs, Ipswich, MA, USA)
overnight at 37°C in 50 μl of the buffers recommended by the
suppliers. Subsequently, the enzymes were inactivated at 65°C for
20 min and the levels of digested DNA were determined by
electrophoresis.
For PCR analysis, 0.5 μl (5 ng) genomic DNA digested
with each enzyme was used in a 10 μl reaction mixture containing
0.25 units Ex-Taq DNA polymerase (Takara Bio Inc., Shiga,
Japan), 4 nmol dNTP (Takara Bio Inc.) and 10 nmol each primer
[Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China] in 5 μl 2×
GC Buffer I or 2× GC Buffer II (Takara Bio Inc.). The thermal
cycling parameters were recommend by Yamada et al (11). The amplified products were
electrophoresed on a 2% agarose gel, stained with Goldview nucleic
acid stain (SBS Genetech, Beijing, China) and visualized by the
Molecular Imager Gel Doc XR+ System (Bio-Rad, Hercules, CA
USA).
Bisulfite genomic sequencing
Human genomic DNA (2 μg) from PBLs was treated with
sodium bisulfite according to the standard procedure. One-tenth of
the bisulfite-treated DNA was used for PCR in a 50 μl reaction
mixture, 10× PCR Buffer [100 mM Tris-HCl (pH 8.8), 500 mM KCl, 15
mM MgCl2 and 0.8% (v/v) Nonidet P40], 10 nmol each dNTP,
10 nmol each primer and 4 units Ex-Taq DNA polymerase [all
Sangon Biotech (Shanghai) Co., Ltd.]. The primer sequences are
described in Table I. The
amplified products were subsequently cloned into a pUC18-T vector
[Sangon Biotech (Shanghai) Co., Ltd.] and sequenced using the
Applied Biosystem 3730 DNA Analyzer (Life Technologies, Carlsbad,
CA, USA). The results were further analyzed using the BDPC DNA
methylation analysis platform (http://services.ibc.uni-stuttgart.de/BDPC/BISMA/).
 | Table ICGI DNA methylation statuses as
determined by bisulfite genomic sequencing. |
Table I
CGI DNA methylation statuses as
determined by bisulfite genomic sequencing.
| Methylation using
bisulfite genomic sequencing | Methylation using
HM-PCR | | |
|---|
|
|
| | |
|---|
| CGI no. | Chinese | Japanese | Chinese | Japanese | Bisulfite genomic
sequencing primers | CGI-linked
genes |
|---|
| 14 | Unmethylation | | Unmethylation | Unmethylation |
5′-GTAGATYGTGTAATTTTATTTATTAGTTAG-3′
5′-CAAAACTACRAAATTCCCAAAC-3′ | ADAM
metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5) |
| 38 | Unmethylation | | Unmethylation | Unmethylation |
5′-GAGTTTGTGGGATTTTGTAGTGAGT -3′
5′-CATCRCCTATCACCTAAACCC-3′ | Regulator of
calcineurin 1 (RCAN1) |
| 41 | Incomplete
methylation | | Incomplete
methylation | Unmethylation |
5′-GGAGGTYGTTTTAGAAAGTTGAGA -3′
5′-CAACCCCAACTTCCTCTACTCC-3′ | Runt-related
transcription factor 1 (RUNX1) |
| 75 | Unmethylation | | Unmethylation | Complete
methylation |
5′-GGGATAAYGATATTTTTTGGGG-3′
5′-CCATACCRACTATTCTTTATTACATTC-3′ | PR domain zinc
finger protein 15 (PRDM15) |
| 103 | Complete
methylation | Composite
methylation | | |
5′-GTAGTTGGGATTATAGTTATATGTTATTATG-3′
5′-CCTCTAAATCCTTATCCCAAAAC-3′ | ES1 protein
homolog, mitochondrial isoform Ib precursor |
| 109 | Incomplete
methylation | | Incomplete
methylation | Unmethylation |
5′-GATGGTTTYGYGGGGTTAGG-3′
5′-RCCCTACAACAACACCRAAACC-3′ | Protein C21orf2
isoform 4 (C21orf2) |
| 129 | Incomplete
methylation | | Incomplete
methylation | Complete
methylation |
5′-GTTTGAGGTTGGTTTAGGTTTTG-3′
5′-CATCTCCCRAATATAAAACTTACTCC-3′ | Folate transporter
1 isoform 3 |
| 133 | Complete
methylation | | Complete
methylation | Complete
methylation |
5′-TATGGTGGTAGGTAAGAGAGTATGTG-3′
5′-CCCRCAAACCCTAAATCTTAAC-3′ | Folate transporter
1 isoform 3 |
| 134 | Incomplete
methylation | | Incomplete
methylation | Complete
methylation |
5′-AAGATTTTGTAGTTGTAAGTGGTGTAG-3′
5′-CCAACTAAATACATATTCTTCCTTCTC-3′ | Poly (rC)-binding
protein 3 (PCBP3) |
| 46 | Complete
methylation | | Complete
methylation | Complete
methylation |
5′-GATATTTTATATTTGTAGGGTTAGTGGA-3′
5′-CAAAACCCCAATCAATCACAC-3′ | Pericentrin
(PCNT) |
Statistical analysis
Statistical analyses were performed using SPSS 10.0
software (SPSS Inc., Chicago, IL, USA). Comparisons between two
groups were made using χ2 tests. P<0.05 was
considered to indicate a statistically significant difference.
Results
CGI sequence alignment data from the BGI
YH database
There are a total of 149 CGIs in chromosome 21q,
according to criteria outlined by Yamada et al (11). The alignment data from the BGI YH
database showed that 87% of CGIs (130 of 149) were >80% matched;
13.42% were a perfect match (20 of 149), 22.14% were >95%
matched (33 of 149), 21.48% were >90% matched (32 of 149),
16.11% were >85% matched (24 of 149) and 14.09% were >85%
matched (21 of 149). A match of <80% accounted for 13% of total
CGIs (19 of 149); 2.01% were >75% matched (3 of 149), 8.05% were
>70% matched (12 of 149), 0.67% were >65% matched (1 of 149),
1.34% were >60% matched (2 of 149) and 0.67% were >55%
matched (1 of 149). The primers of the 148 CGIs were obtained from
the experimental design of Yamada et al (11). The alignment data indicated that
98% (145 of 148) of the primers were a perfect match, two pairs of
primers (CGI nos. 27 and 75) were a 50% match and one pair of
primers (CGI no. 90) was not a match. New primers for CGI nos. 27,
75 and 90 were designed using Primer 3 (v0.4.0; http://primer3.wi.mit.edu/) as follows: Forward:
5′-CTCTCACCGCCGCAAGTCGGTCGC-3′ and reverse:
5′-CGTTGTTGGGGAACTTTTACTGTG-3′ for CGI no. 27; forward:
5′-CAGCTCAAGAATGCACTGCATTT-3′ and reverse:
5′-AGTCAAAACCCGGCTGGATTTCC-3′ for CGI no. 75; and forward:
5′-GTATGTGCCACCAAA TGATTATTCCT-3′ and reverse: 5′-ACTCACTCTCCTAAC
TTGAAGTTTTC-3′ for CGI no. 90.
HpaII-McrBC PCR assay to evaluate 21q CGI
allelic methylation status
The electrophoresed images of genomic DNA and DNA
products digested by McrBC, HpaII or HhaI
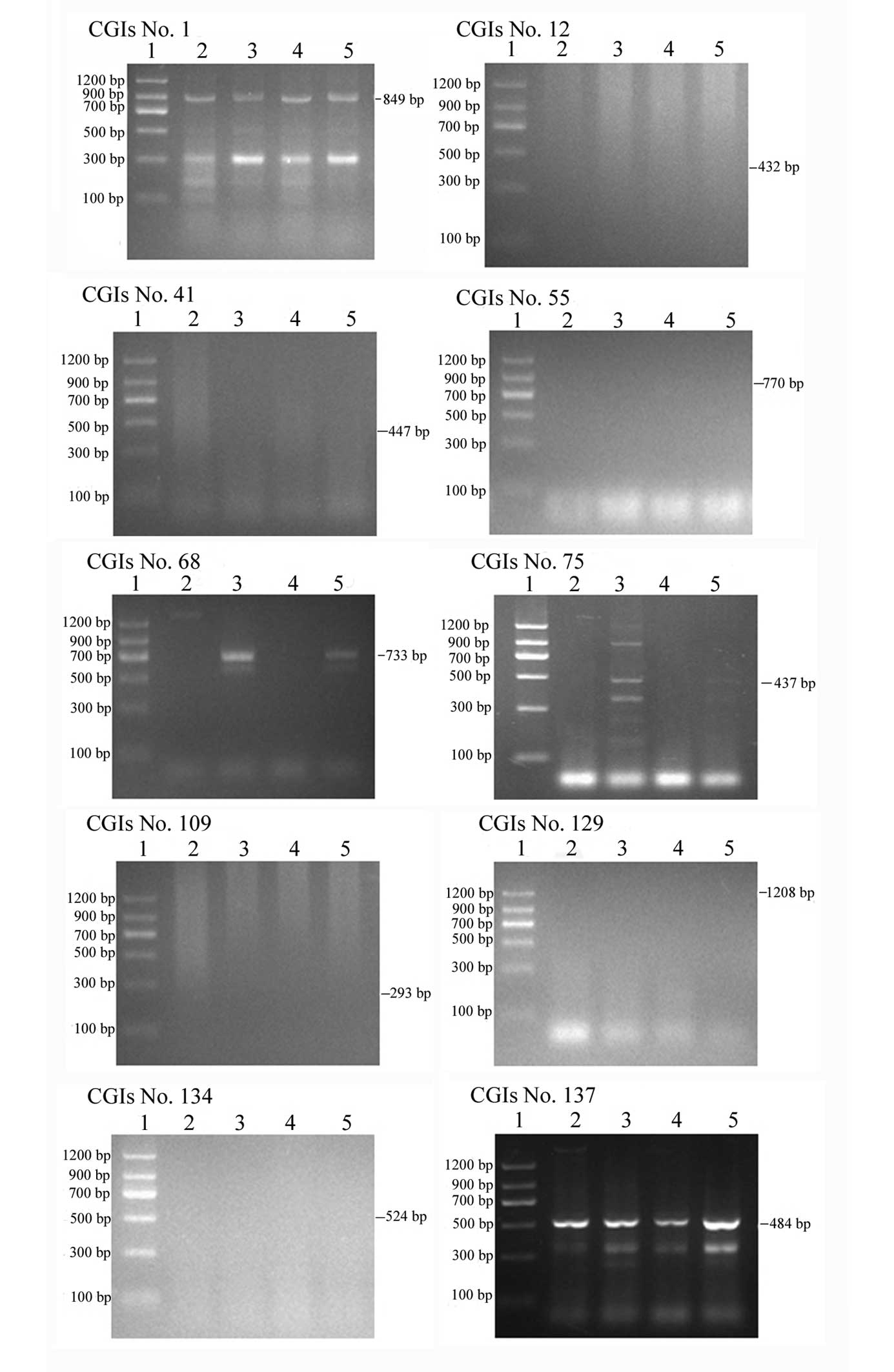
enzymes, respectively, are shown in Fig. 1. When the genomic DNA were
completely digested by the enzyme, the DNA products appeared as
smeared bands in gel electrophoresis. Using the heavy
methyl-polymerase chain reaction (HM-PCR) assay, results showed
that there was almost no difference in the DNA methylation status
of 21q CGIs among individuals with trisomy 21 and the control, as
shown in Table II. A total of 148
CGIs in 21q were screened, including 102 null methylation, 26
complete methylation, 7 composite methylation and 13 incomplete
methylation. However, 3 null methylation CGIs (nos. 12, 41 and
109), 2 complete methylation (nos. 129 and 134) and 1 composite
methylation (no. 55) in Japanese patients were all incomplete
methylation in Chinese patients. In addition, 1 incomplete
methylation CGI (no. 68) and 1 complete methylation CGI (no. 75) in
Japanese patients were also incomplete methylation in Chinese
patients. Finally, 1 complete methylation CGIs (nos. 1 and 137) in
Japanese patients were composite methylation in Chinese. In total,
there were 10 CGIs that showed varying DNA methylation statuses
among Japanese and Chinese patients, as presented in Fig. 2 and Table II.
| Table IIVariations in DNA methylation
statuses between Chinese and Japanese patients determined using
HM-PCR. |
Table II
Variations in DNA methylation
statuses between Chinese and Japanese patients determined using
HM-PCR.
| Methylation status
detected using HM-PCR | Methylation status
detected using bisulfite genomic sequencing | |
|---|
|
|
| |
|---|
| CGI no. | Chinese | Japanese | Chinese | Japanese | CGI-linked
genes |
|---|
| 1 | Composite
methylation | Complete
methylation | | | LON peptidase
N-terminal domain and ring finger 2 (LONRF2) |
| 12 | Incomplete
methylation | Unmethylation | | | ADAM
metallopeptidase with thrombospondin type 1 motif 1 (ADAMTS1) |
| 41 | Incomplete
methylation | Unmethylation | Incomplete
methylation | | Runt-related
transcription factor 1 (RUNX1) |
| 55 | Incomplete
methylation | Composite
methylation | | Composite
methylation | Holocarboxylase
synthetase (HLCS) |
| 68 | Unmethylation | Incomplete
methylation | | | Tryptophan rich
basic protein (WRB) |
| 75 | Unmethylation | Complete
methylation | Unmethylation | | PR domain zinc
finger protein 15 (PRDM15) |
| 109 | Incomplete
methylation | Unmethylation | Incomplete
methylation | | Protein C21orf2
isoform 4 |
| 129 | Incomplete
methylation | Complete
methylation | Incomplete
methylation | | Folate transporter
1 isoform 3 (SLC19A3) |
| 134 | Incomplete
methylation | Complete
methylation | Incomplete
methylation | | Poly (rC)-binding
protein 3 (PCBP3) |
| 137 | Composite
methylation | Complete
methylation | | Complete
methylation | Collagen α-1(VI)
chain precursor (COL6A1) |
Bisulfite genomic sequencing to confirm
allelic methylation status
Nine CGIs (nos. 14, 38, 41, 75, 109, 129, 133, 134
and 146) were selected to validate the HM-PCR assay data using
bisulfite sequencing in trisomy 21 and the control, as shown in
Table I. These validations were
determined with bisulfite sequencing, which showed that CGI nos.
41, 109, 129 and 134 were incomplete methylation, CGI nos. 14, 38
and 75 were null methylation and CGI nos. 133 and 146 were complete
methylation in Chinese patients, as shown in Fig. 3; these data validate the HM-PCR
results. The methylation status of CGI no. 103 was detected by
bisulfite sequencing, as it was short of the HpaII and
HhaI recognition sites. The composite methylation CGI no.
103 in Japanese patients was complete methylation in Chinese
patients, as shown in Table I and
Fig. 3. Furthermore, the various
methylation statuses of CpG dinucleotides in 5 CGIs between trisomy
21 and the control were found, including 5 sites in CGI no. 14, 6
sites in CGI no. 75, 14 sites in CGI no. 109, 1 site in CGI no. 134
and 3 sites in CGI no. 146. A total of 29 CpG dinucleotides
presented allele-specific DNA methylation, as shown in Fig. 3.
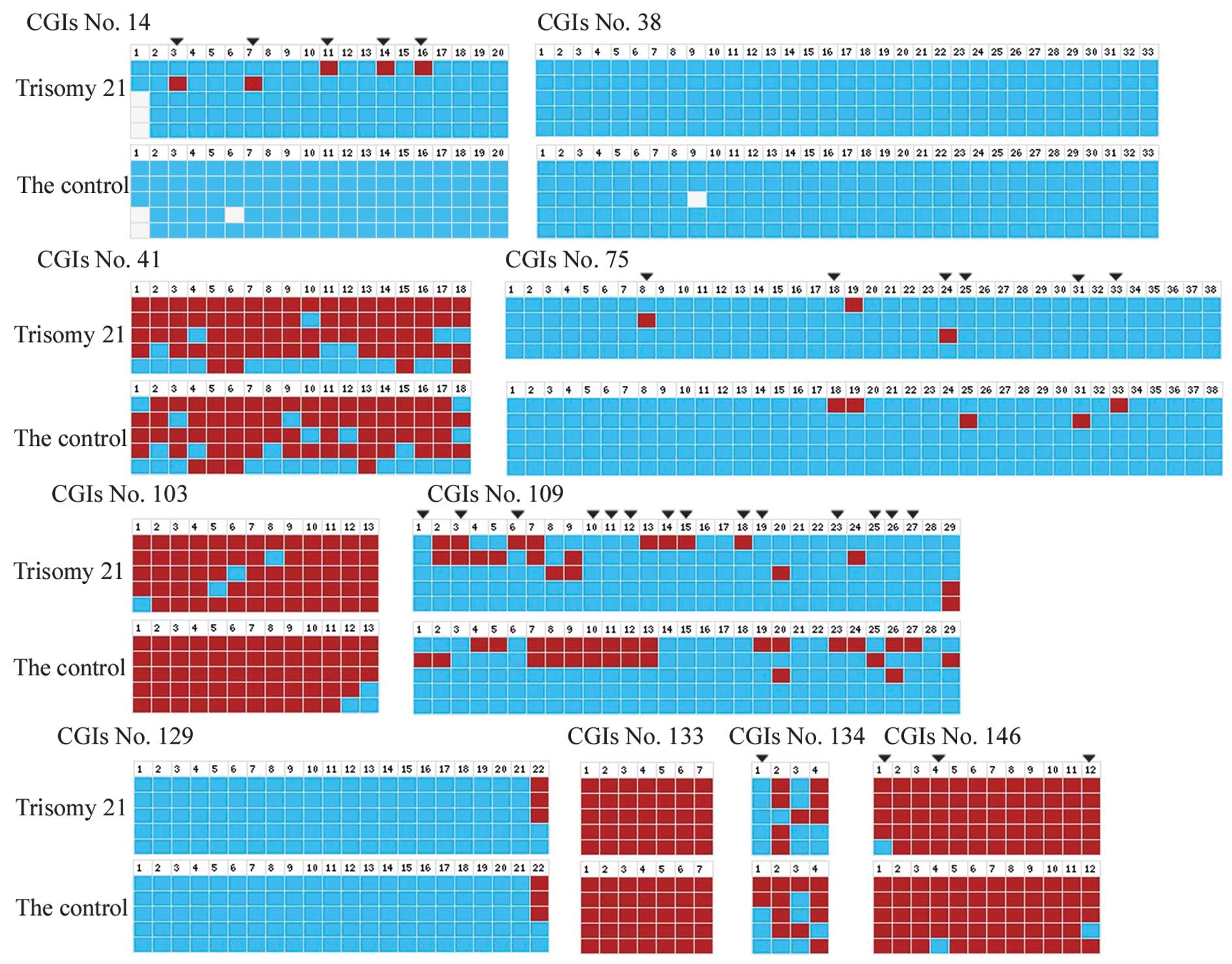 | Figure 3Methylation status of ten CGIs on
chromosome 21q as determined by bisulfite genomic sequencing,
defined as methylated (red), unmethylated (blue) and unknown
(white). The different methylation statuses in CpG dinucleotide
sites between trisomy 21 and the controls are indicated by downward
arrows. Image depicts the various methylation CpG dinucleotide
sites in CGI no. 14 (nos. 3, 7, 11, 14 and 16), 75 (nos. 8, 18, 24,
25, 31 and 33), 109 (nos. 1, 3, 6, 10, 11, 12, 14, 15, 18, 19, 23,
25, 26 and 27), 134 (no. 1) and 146 (no. 1, 4 and 12). CGI, CpG
island. |
Discussion
Individuals with DS have an additional chromosome
21, which is associated with the gene-dosage effect and a wide
spectrum of phenotypic consequences, including life-threatening
complications, clinically significant alteration of life course
(e.g., mental retardation) and dysmorphic physical features
(12). The mechanisms of gene
regulation, include function of conserved nongenic regions,
microRNA activities, RNA editing and DNA methylation. DS with a CHD
is associated with a global hypomethylation status (13). DNA methylation is a possible
mechanism of gene expression alteration, which may contribute to
various abnormalities. Chango et al (14) used a combination of
methylation-sensitive arbitrarily primed PCR and quantitation of
DNA fragments to find six fragments that were hypermethylated in
PBLs from eight individuals with DS, compared with eight normal
controls. Kerkel et al (10) observed that 8 genes had different
methylation status between the DS patients and normal controls. One
of the 8 genes is named SUMO3 and is located on chromosome 21. The
current observations are consistent with this data. There were
differences in the DNA methylation status of CpG dinucleotide sites
in 21q CGIs (nos. 14, 75, 109, 134 and 146) among individuals with
trisomy 21 and the control. Molecular analysis reveals that the
21q22.1-q22.3 region, also known as the DS critical region (DSCR),
appears to contain the gene or genes responsible for the CHD
observed in DS (15–17). Altered DNA methylation in 21q may
be constitutively silenced overexpressed genes in DS (10). Noteworthy gene candidates for
specific dysfunctions in DS are already emerging from these
research data. In the present study, CGI no. 75 was linked to
PRDM15, which is a candidate gene for a particular phenotype
of DS or bipolar affective disorder (18). CGI no. 14 was associated with
ADAMTS5, which is a protease involved in regulating
aggrecanase activity in cartilage; deletion of active ADAMTS5
prevents cartilage degradation (19,20).
CGI no. 109 was associated with C21orf2, which is involved
in the regulation of cell morphology and cytoskeletal organization
(21). CGI no. 134 was associated
with PCBP3, which is associated with frontotemporal dementia
and hypothesized to participate in mRNA metabolic processes
(22,23). CGI no. 146 was associated with
PCNT, which may be important in preventing premature
centrosome splitting during interphase by inhibition of NEK2 kinase
activity at the centrosome (24).
The methylation alteration may be associated with the DS phenotype,
as insights from investigating DS as a model system have shed light
on potential epigenetic biomarkers for noninvasive prenatal
diagnosis in the general population.
The fetal-specific DNA methylation ratio permits
noninvasive prenatal diagnosis of trisomy 21 by analyzing
fetal-specific DMRs in free fetal DNA of the maternal circulation
during pregnancy (25–28). A few differentially methylated
sequences are fetal DNA methylation markers, including HLCS
and DSCR4 located at chromosome 21, RASSF1A located
at chromosome 3 and ZFY located at chromosome Y (29–31).
However, these markers were not specific for individuals with DS.
In the present study, methylation alteration in DS may represent a
specific epigenetic biomarker for future prenatal diagnosis;
however, DNA methylation must be further verified in fetuses with
DS.
DNA methylation in DS individuals did not change
significantly in comparison with the controls (14). In the present study, there was no
significant difference in 21q CGIs in Chinese patients, as
determined using HM-PCR; this result was in accordance with
observations from Kerkel et al (10) of no significant difference between
normal and DS samples. While the different methylation statuses of
CpG dinucleotide sites between the normal and DS do not cause the
difference in the CGIs’ methylation status as screened by HM PCR
assays, because CGIs comprise numerous CpG dinucleotide sites only
a few altered methylation levels are likely to not affect HM PCR
results (11). Varying DNA
methylation statuses of CGIs in 21q existed among Japanese and
Chinese patients. According to the results of alignment in the BGI
YH database, data showed that 87% of CGIs (130 of 149) were >80%
matched and 149 CGIs were feasible for the analysis of Chinese DNA
sequences in the HM-PCR assay (32). In total, 149 CGIs in 21q were
screened, including 102 null methylation, 26 complete methylation,
7 composite methylation and 13 incomplete methylation. There were
11 DNA methylation statuses of CGIs, no. 1, 12, 41, 55, 68, 75,
103, 109, 129, 134 and 137, among Japanese and Chinese patients.
The racial disparities in DNA methylation patterns of differing
ethnic groups implicate the probable role of molecular markers in
determining an individual’s susceptibility to disease. Racial
disparities in DNA methylation patterns have been found in prostate
cancer, endometrial carcinoma, breast cancer and laryngeal cancer
and, in addition, are associated with racial difference in the
cancer prognosis and survival rate (33–37).
CGI no. 12 is associated with ADAMTS1, which is a protease
involved in extracellular matrix proteolysis and antiangiogenesis
and is involved in ischemia-induced retinal neovascularization,
overexpressed in neurodegenerative disorders and downregulated in
breast carcinomas (38–41). CGI no. 41 is associated with
RUNX1, which is linked to a poor outcome of acute
lymphoblastic leukemia and susceptibility to autoimmune disease;
emergence of the RUNX1 mutations was detected in advanced
chronic myelogenous leukemias with acquired trisomy 21 (42–46).
CGI no. 55 is associated with HLCS and a lack of HLCS
may cause multiple carboxylase deficiency (47). CGI no. 68 is associated with
WRB, which is a conserved tryptophan-rich motif in the
membrane-proximal region of the HIV-1 gp41 ectodomain and is
important for Env-mediated fusion and virus infectivity (46,48).
CGI no. 129 is associated with SLC19A1, a transporter for
the intake of folate (49). CGI
no. 137 is associated with COL6A1, depletion of which is a
cause of Bethlem myopathy and Ullrich congenital muscular dystrophy
(50).
In conclusion, the different DNA methylation status
of 21q CGIs status between Chinese and Japanese individuals, and
the same DNA methylation status detected by HM-PCR between Chinese
individuals with DS and the control indicates that DNA methylation
is likely to be an epigenetic marker for distinguishing
ethnicities. The different DNA methylation status of CpG
dinucleotides between individuals with DS and the control may
contribute to the DS complex phenotypes and be a potential
epigenetic marker for diagnosing trisomy 21.
Acknowledgements
The authors would like to thank Dr Lin Zou and Dr
Jing Yang (Center for Clinical Molecular Medicine, Children’s
Hospital of Chongqing Medical University) for case collection. The
present study was supported by grants from the Chongqing Key
Laboratory of Birth Defects and Reproductive Health (grant no.
0901) and the Natural Science Foundation Project of CQ CSTC (grant
no. 2009BA5082).
References
|
1
|
Huether CA, Gummere GR, Hook EB, et al:
Down’s syndrome: percentage reporting on birth certificates and
single year maternal age risk rates for Ohio 1970–79: comparison
with upstate New York data. Am J Public Health. 71:1367–1372.
1981.
|
|
2
|
Karlsson B, Gustafsson J, Hedov G,
Ivarsson SA and Anneren G: Thyroid dysfunction in Down’s syndrome:
relation to age and thyroid autoimmunity. Arch Dis Child.
79:242–245. 1998.
|
|
3
|
Nespoli L, Burgio GR, Ugazio AG and
Maccario R: Immunological features of Down’s syndrome: a review. J
Intellect Disabil Res. 37:543–551. 1993.
|
|
4
|
Yang Q, Rasmussen SA and Friedman JM:
Mortality associated with Down’s syndrome in the USA from 1983 to
1997: a population-based study. Lancet. 359:1019–1025. 2002.
|
|
5
|
Caputo AR, Wagner RS, Reynolds DR, Guo SQ
and Goel AK: Down syndrome. Clinical review of ocular features.
Clin Pediatr (Phila). 28:355–358. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shott SR, Joseph A and Heithaus D: Hearing
loss in children with Down syndrome. Int J Pediatr
Otorhinolaryngol. 61:199–205. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lejeune J, Turpin R and Gautier M:
Mongolism; a chromosomal disease (trisomy). Bull Acad Natl Med.
143:256–265. 1959.(In French).
|
|
8
|
Daura-Oller E, Cabre M, Montero MA,
Paternain JL and Romeu A: Specific gene hypomethylation and cancer:
new insights into coding region feature trends. Bioinformation.
3:340–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smolarek I, Wyszko E, Barciszewska AM, et
al: Global DNA methylation changes in blood of patients with
essential hypertension. Med Sci Monit. 16:CR149–CR155.
2010.PubMed/NCBI
|
|
10
|
Kerkel K, Schupf N, Hatta K, et al:
Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet.
6:e10012122010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamada Y, Watanabe H, Miura F, et al: A
comprehensive analysis of allelic methylation status of CpG islands
on human chromosome 21q. Genome Res. 14:247–266. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Patterson D: Genetic mechanisms involved
in the phenotype of Down syndrome. Ment Retard Dev Disabil Res Rev.
13:199–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Obermann-Borst SA, van Driel LM, Helbing
WA, et al: Congenital heart defects and biomarkers of methylation
in children: a case-control study. Eur J Clin Invest. 41:143–150.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chango A, Abdennebi-Najar L, Tessier F, et
al: Quantitative methylation-sensitive arbitrarily primed PCR
method to determine differential genomic DNA methylation in Down
Syndrome. Biochem Biophys Res Commun. 349:492–496. 2006. View Article : Google Scholar
|
|
15
|
Laue L, Chan WY, Hsueh AJ, et al: Genetic
heterogeneity of constitutively activating mutations of the human
luteinizing hormone receptor in familial male-limited precocious
puberty. Proc Natl Acad Sci USA. 92:1906–1910. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arron JR, Winslow MM, Polleri A, et al:
NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on
chromosome 21. Nature. 441:595–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ronan A, Fagan K, Christie L, et al:
Familial 4.3 Mb duplication of 21q22 sheds new light on the Down
syndrome critical region. J Med Genet. 44:448–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shibuya K, Kudoh J, Okui M and Shimizu N:
Identification of a novel zinc finger protein gene (ZNF298) in the
GAP2 of human chromosome 21q. Biochem Biophys Res Commun.
332:557–568. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vankemmelbeke MN, Holen I, Wilson AG, et
al: Expression and activity of ADAMTS-5 in synovium. Eur J Biochem.
268:1259–1268. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Glasson SS, Askew R, Sheppard B, et al:
Deletion of active ADAMTS5 prevents cartilage degradation in a
murine model of osteoarthritis. Nature. 434:644–648. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bai SW, Herrera-Abreu MT, Rohn JL, et al:
Identification and characterization of a set of conserved and new
regulators of cytoskeletal organization, cell morphology and
migration. BMC Biol. 9:542011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiledjian M, Wang X and Liebhaber SA:
Identification of two KH domain proteins in the alpha-globin mRNP
stability complex. EMBO J. 14:4357–4364. 1995.PubMed/NCBI
|
|
23
|
Wang Y, Gao L, Tse SW and Andreadis A:
Heterogeneous nuclear ribonucleoprotein E3 modestly activates
splicing of tau exon 10 via its proximal downstream intron, a
hotspot for frontotemporal dementia mutations. Gene. 451:23–31.
2010. View Article : Google Scholar
|
|
24
|
Matsuo K, Nishimura T, Hayakawa A, Ono Y
and Takahashi M: Involvement of a centrosomal protein kendrin in
the maintenance of centrosome cohesion by modulating Nek2A kinase
activity. Biochem Biophys Res Commun. 398:217–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Papageorgiou EA, Karagrigoriou A, Tsaliki
E, et al: Fetal-specific DNA methylation ratio permits noninvasive
prenatal diagnosis of trisomy 21. Nat Med. 17:510–513. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Papageorgiou EA, Fiegler H, Rakyan V, et
al: Sites of differential DNA methylation between placenta and
peripheral blood: molecular markers for noninvasive prenatal
diagnosis of aneuploidies. Am J Pathol. 174:1609–1618. 2009.
View Article : Google Scholar
|
|
27
|
Old RW, Crea F, Puszyk W and Hultén MA:
Candidate epigenetic biomarkers for non-invasive prenatal diagnosis
of Down syndrome. Reprod Biomed Online. 15:227–235. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan KC, Ding C, Gerovassili A, et al:
Hypermethylated RASSF1A in maternal plasma: A universal fetal DNA
marker that improves the reliability of noninvasive prenatal
diagnosis. Clin Chem. 52:2211–2218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chim SS, Jin S, Lee TY, et al: Systematic
search for placental DNA-methylation markers on chromosome 21:
toward a maternal plasma-based epigenetic test for fetal trisomy
21. Clin Chem. 54:500–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Du Y, Zhang J, Wang H, et al:
Hypomethylated DSCR4 is a placenta-derived epigenetic marker for
trisomy 21. Prenat Diagn. 31:207–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tong YK, Jin S, Chiu RW, et al:
Noninvasive prenatal detection of trisomy 21 by an
epigenetic-genetic chromosome-dosage approach. Clin Chem. 56:90–98.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Wang W, Li R, et al: The diploid
genome sequence of an Asian individual. Nature. 456:60–65. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Woodson K, Hayes R, Wideroff L, Villaruz L
and Tangrea J: Hypermethylation of GSTP1, CD44, and E-cadherin
genes in prostate cancer among US Blacks and Whites. Prostate.
55:199–205. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Woodson K, Hanson J and Tangrea J: A
survey of gene-specific methylation in human prostate cancer among
black and white men. Cancer Lett. 205:181–188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adkins RM, Krushkal J, Tylavsky FA and
Thomas F: Racial differences in gene-specific DNA methylation
levels are present at birth. Birth Defects Res A Clin Mol Teratol.
91:728–736. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nielsen DA, Hamon S, Yuferov V, et al:
Ethnic diversity of DNA methylation in the OPRM1 promoter region in
lymphocytes of heroin addicts. Hum Genet. 127:639–649. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dumitrescu RG, Marian C, Krishnan SS, et
al: Familial and racial determinants of tumour suppressor genes
promoter hypermethylation in breast tissues from healthy women. J
Cell Mol Med. 14:1468–1475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bevitt DJ, Mohamed J, Catterall JB, et al:
Expression of ADAMTS metalloproteinases in the retinal pigment
epithelium derived cell line ARPE-19: transcriptional regulation by
TNFalpha. Biochim Biophys Acta. 1626:83–91. 2003. View Article : Google Scholar
|
|
39
|
Porter S, Scott SD, Sassoon EM, et al:
Dysregulated expression of adamalysin-thrombospondin genes in human
breast carcinoma. Clin Cancer Res. 10:2429–2440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu Z, Yu Y and Duh EJ: Vascular
endothelial growth factor upregulates expression of ADAMTS1 in
endothelial cells through protein kinase C signaling. Invest
Ophthalmol Vis Sci. 47:4059–4066. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Miguel RF, Pollak A and Lubec G:
Metalloproteinase ADAMTS-1 but not ADAMTS-5 is manifold
overexpressed in neurodegenerative disorders as Down syndrome,
Alzheimer’s and Pick’s disease. Brain Res Mol Brain Res. 133:1–5.
2005.
|
|
42
|
Helms C, Cao L, Krueger JG, et al: A
putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is
associated with susceptibility to psoriasis. Nat Genet. 35:349–356.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu S, Shen T, Huynh L, et al: Interplay
of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid
leukemia. Cancer Res. 65:1277–1284. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Roche-Lestienne C, Deluche L, Corm S, et
al: RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in
BCR-ABL+ leukemias are frequently associated with
secondary trisomy 21 and may contribute to clonal evolution and
imatinib resistance. Blood. 111:3735–3741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Strefford JC, van Delft FW, Robinson HM,
et al: Complex genomic alterations and gene expression in acute
lymphoblastic leukemia with intrachromosomal amplification of
chromosome 21. Proc Natl Acad Sci USA. 103:8167–8172. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang Y, Strissel P, Strick R, et al:
Genomic DNA breakpoints in AML1/RUNX1 and ETO cluster with
topoisomerase II DNA cleavage and DNase I hypersensitive sites in
t(8;21) leukemia. Proc Natl Acad Sci USA. 99:3070–3075. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tang NL, Hui J, Yong CK, et al: A genomic
approach to mutation analysis of holocarboxylase synthetase gene in
three Chinese patients with late-onset holocarboxylase synthetase
deficiency. Clin Biochem. 36:145–149. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Salzwedel K, West JT and Hunter E: A
conserved tryptophan-rich motif in the membrane-proximal region of
the human immunodeficiency virus type 1 gp41 ectodomain is
important for Env-mediated fusion and virus infectivity. J Virol.
73:2469–2480. 1999.
|
|
49
|
Börjel AK, Yngve A, Sjöström M and Nilsson
TK: Novel mutations in the 5′-UTR of the FOLR1 gene. Clin Chem Lab
Med. 44:161–167. 2006.
|
|
50
|
Lampe AK, Dunn DM, von Niederhausern AC,
et al: Automated genomic sequence analysis of the three collagen VI
genes: applications to Ullrich congenital muscular dystrophy and
Bethlem myopathy. J Med Genet. 42:108–120. 2005. View Article : Google Scholar
|