Introduction
Diabetic retinopathy is one of the most common
complications in patients with diabetes and affects ~75% of them
within 15 years of disease onset. In a number of patients, diabetic
retinopathy may progress into proliferative retinopathy, which is
characterized by the growth of new blood vessels on the surface of
the retina. Hyperglycemia is the primary pathogenic factor in the
development of diabetic complications (1). Retinal capillary damage from high
glucose-induced pericyte loss and alterations in retinal
hemodynamics results in a hypoxic state in the early and late
stages of diabetic retinopathy (2,3). It
has been previously established that the signaling effects of
hyperglycemia and hypoxia contribute to the progression of diabetic
retinopathy (4).
The activation of protein kinase C (PKC) is a key
feature of diabetes mellitus. PKC translocation to the plasma
membrane is considered as a hallmark of PKC activation and is
frequently used to measure PKC isoform activation in cells.
Increased PKC activation has been associated with changes in blood
flow, basement membrane thickening, extracellular matrix expansion,
increases in vascular permeability and abnormal angiogenesis, and
has been implicated in the pathogenesis of diabetic retinopathy
(5). PKC is a complex
serine/threonine kinase family. At present, three groups and 12
isoforms of PKC have been determined in the following types of
tissue: i) Classic or typical PKCs, including α, βI, βII and γ,
which are calcium- and diacylglycerol (DAG)-dependent; ii) novel
PKCs, including δ, ɛ, η and θ, which are calcium-independent and
DAG-dependent; and iii) atypical PKCs, including ζ and ι/λ, which
are calcium and DAG-independent and are only activated by
phosphatidylserine (6). The
distribution of different PKC isoforms is commonly tissue- and
species-dependent. In addition, various types of PKC isoforms may
be expressed in the same cells, but with distinct biological
functions. Alterations in the activities of distinct PKC isoforms
have been linked to the development of macro- and microvascular
complications observed in patients with diabetes (7). In bovine retinal endothelial cells,
PKC α, βI, βII, δ, ɛ and ζ, representing all of the three PKC
subgroups, have been detected (8).
However, in primary cultured human retinal endothelial cells
(HRECs) the distribution of PKC isoforms has not been fully
elucidated. Furthermore, the association between translocation of
PKC isoforms and proliferation of HRECs remains to be
elucidated.
The results of a previous study by our group
identified a significant increase in the proliferation of HRECs at
moderately high glucose concentrations and hypoxic conditions
(9). Notably, high glucose
concentrations alone did not significantly induce proliferation of
HRECs. Therefore, the present study aimed to examine the
subcellular distribution of PKC isoforms in cultured HRECs and
analyze the translocation of PKC isoforms under the abovementioned
glucose and hypoxic conditions.
Materials and methods
Culture of HRECs
HRECs were isolated from tissue a patient undergoing
corneal transfer surgery at Zhongshan Ophthalmic Center, Sun
Yat-Sen University (Guangzhou, China) as previously described
(10). Briefly, cells were grown
in human endothelial serum free-medium (Gibco-BRL, Carlsbad, CA,
USA) supplemented with 10% fetal bovine serum (Thermo Fisher
Scientific, Hyclone, Logan, UT, USA), 5 μg/l β-endothelial cell
growth factor (β-ECGF; R&D Systems Inc., Minneapolis, MN, USA)
and 1% (v/v) penicillin-streptomycin (Gibco-BRL, Grand Island, NY,
USA) at 37°C in a 5% CO2 and 95% air atmosphere. Cells
were digested with Trypsinase-EDTA (Sigma-Aldrich, St. Louis, MO,
USA) and subcultured in 25 cm2 culture flasks for cell
fractionation or 96-well plates for proliferation assay under the
appropriate assay conditions. The number of live cells were counted
using a blood counting instrument (Buerker; Brand GmbH and Co. KG,
Wertheim, Germany) following 0.4% trypan blue staining. Cells at
passages three to six were used in the experiments. The experiments
adhered to the tenets of the Declaration of Helsinki for research
involving human subjects. Ethical approval was obtained from the
Ethics Committee of Zhongshan Ophthalmic Center and informed
consent was obtained from all the donor patient.
HREC proliferation assay
HRECs were trypsinized (trypsinase-EDTA) and seeded
in 96-well plates (~5,000 cells/well). Following overnight
incubation, cells were transferred into serum-free medium (without
β-ECGF) and were further incubated for 24 h. The medium was then
replaced with growth medium and cells were further incubated under
one of the following conditions for an additional 48 h: normal
glucose concentration [5 mM D-glucose (Sigma-Aldrich; control
group)]; moderately high glucose (15 mM D-glucose; MG group); high
glucose (30 mM D-glucose; HG group); 150 μM CoCl2
(Sigma-Aldrich; HO group), which is known to trigger chemical
hypoxia in cells, and with or without rottlerin (10 μM; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA; RO group) or Ro 32-0432
(200 nM; Calbiochem, La Jolla, CA, USA; Ro32 group). Equal molar
ratios of mannitol (Sigma-Aldrich) were used as an osmotic control:
15 mM D-mannitol (MM group) and 30 mM D-mannitol (HM group). For
quantitative analysis of cell viability, 10 μl of a cell counting
MTT solution (Sigma, St. Louis, MO, USA) was added to each well and
incubated for 4 h at 37°C in a humidified CO2 incubator.
Absorbance at 490 nm was measured with a microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA). Bromodeoxyuridine (BrdU)
reagent (BrdU Cell Proliferation Assay kit; Millipore, Bedford, MA,
USA) was diluted to 60 μM and added into the wells 24 h following
treatment of the cells under the various conditions. Following
incubation, absorbance was measured at 450 nm.
Cell fractionation
Whole cell lysates
HRECs were washed twice with ice-cold
phosphate-buffered saline (PBS) and lysed in western and
immunoprecipitation cell lysis buffer (Beyotime, Shanghai, China)
with 1 mmol Na3VO4, 1 mmol NaF
(Sigma-Aldrich) and Protease Inhibitor Cocktail Set®
(Calbiochem, Darmstadt, Germany), and incubated for 30 min on ice
then centrifuged at 15,000 × g for 15 min at 4°C. The supernatants
were collected.
Subcellular lysates
HREC lysates were subfractionated into cytosolic and
membrane fractions by adapting the methods previously described
(11). Briefly, HRECs were washed
twice with ice-cold PBS, scraped in lysis buffer A [1 mM
NaHCO3, 5 mM MgCl2·6H2O, 50 mM
Tris-HCl, 10 mM ethylene glycol tetraacetic acid, 2 mM EDTA, 500 μM
4-(2-aminoethyl)-benzenesulfonyl fluoride, 150 nM aprotinin, 1 μM
leupeptin, 1 μM E-46 protease inhibitor] at 4°C, homogenized by
passing through a 26 gauge needle five times, incubated for 15 min
on ice and ultracentrifuged at 100,000 × g for 1 h at 4°C. The
supernatant provided the cytosolic fraction. The pellet was
resuspended in buffer B (buffer A with 1% Triton X-100),
homogenized by passing through a 26 gauge needle five times,
incubated for 15 min on ice and ultracentrifuged at 100,000 × g for
1 h at 4°C. The supernatant provided the membrane fraction. The
protein concentration in all samples were determined by the
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology, Shanghai, China) using bovine serum albumin (BSA) as
the standard.
Western blot analysis
Equal amounts of protein from cell lysates (20–80
μg) or cell fractions of the different treatment groups, along with
appropriate molecular size standards and the rat brain positive
control were separated using 8% SDS-PAGE and blotted onto a
polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA)
for 1.5 h at 100 V using a Bio-Rad transblot apparatus (Bio-Rad,
Hercules, CA, USA). The membrane was blocked for 2 h at room
temperature with tris-buffered saline with Tween 20 (TBST)
containing 5% fat-free milk. Following three washes with TBST, the
membrane was incubated overnight at 4°C with anti-PKC βI, βII
(1:200; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-PKC
α, δ, ɛ and ζ [1:1,000, Cell Signaling Technology, Danvers, MA,
USA], anti-β-actin (1:2,000; β-actin was used as the loading
control of the cytosolic fraction; Cell Signaling Technology)
anti-Na+-K+-ATPase (1:500,
Na+-K+-ATPase was used as the loading control
of the membrane fraction; Cell Signaling Technology) in 5% fat-free
milk or 5% (BSA). The membrane was then washed with TBST and
incubated for 1.5 h at room temperature with a horseradish
peroxidase (HRP)-labeled anti-mouse immunoglobulin G (IgG; 1:2,000,
Cell Signaling Technology) or HRP-labeled anti-rabbit IgG (1:2,000;
Invitrogen Life Technologies, Carlsbad, CA, USA). Antigen detection
was performed with an enhanced chemiluminescence detection system
(Cell Signaling Technology).
Immunofluorescence labeling and confocal
microscopy
HRECs were plated in six-well plates with a
coverslip over each well and incubated in growth medium. Following
overnight incubation, cells were transferred into a serum-free
medium and incubated for 24 h. The medium was then replaced with
different growth media, including normal glucose (5 mM D-glucose),
moderately high glucose (15 mM D-glucose), high glucose (30 mM
D-glucose) and 150 μM CoCl2 and incubated for 48 h. The
cells were then washed twice with ice-cold PBS, fixed with 3.7%
formaldehyde for 15 min at room temperature and the membranes were
permeabilized with 0.1% Triton X-100 for 5 min. Cells were washed
three times with PBS and blocked with 1% goat serum plus 0.1% BSA
in PBS for 1 h at room temperature. Antibodies for immunoblotting
were diluted at 1:100 in blocking solution, added to each coverslip
and incubated for 1 h at 37°C. Following washing three times with
TBST, the secondary fluorescein isothiocyanate-conjugated antibody
was diluted at 1:200 in blocking solution, added to each coverslip,
incubated for 1 h at 37°C in the dark and washed three times with
TBST. A confocal laser scanning image system (FV500; Olympus,
Tokyo, Japan) was used to detect immunofluorescence.
Statistical analysis
All experiments were performed in triplicate.
Results are expressed as the mean ± standard deviation. One-way
analysis of variance was used to analyze all data and the multiple
comparisons were conducted by Tukey’s test. P<0.05 was
considered to indicate a statistically significant difference.
Results
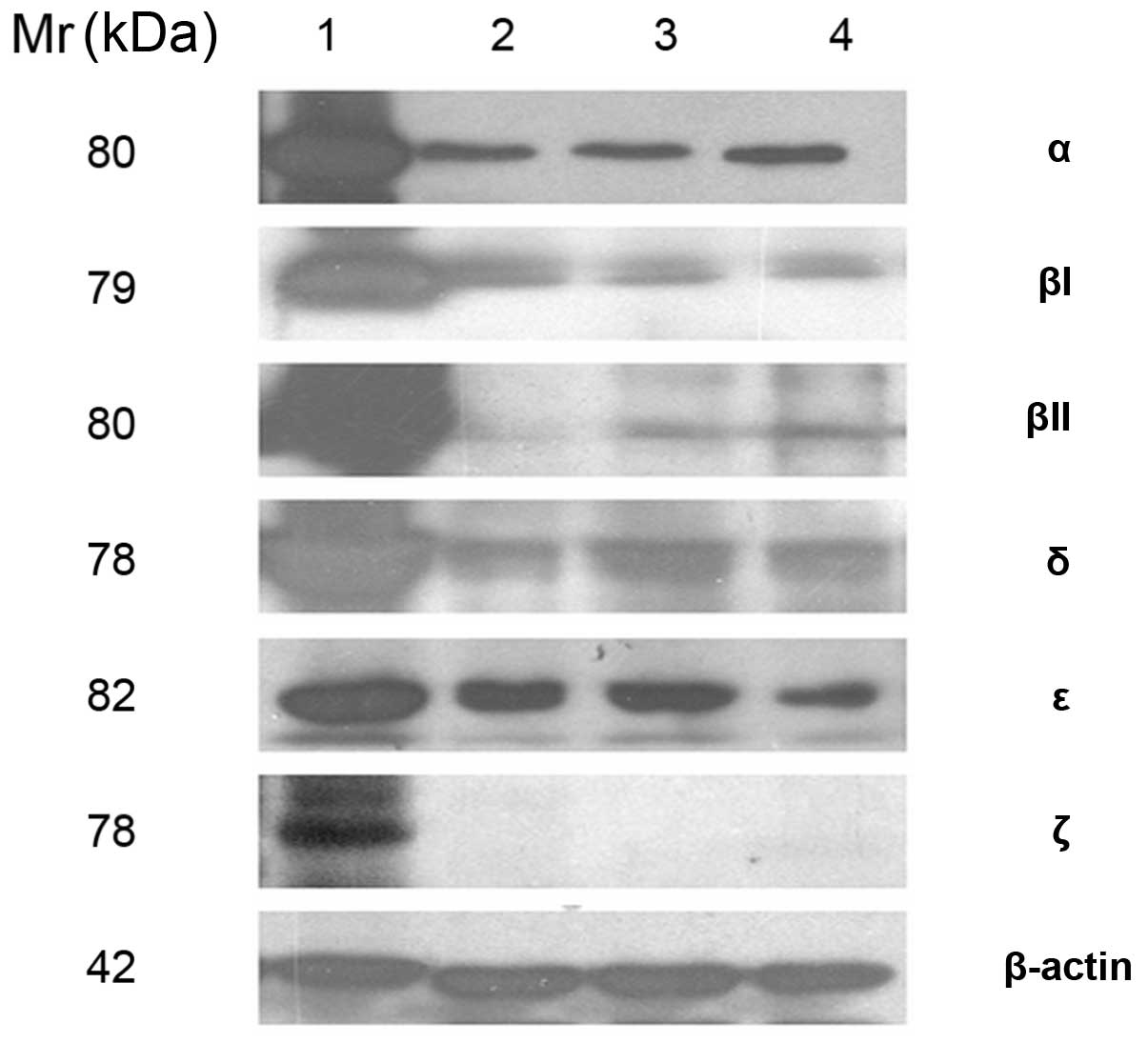
Expression of PKC isoforms in HRECs
As shown in Fig. 1,
PKC isoforms were detected by immunoblotting from total cell lysate
of primary cultured HRECs. The expression of PKC α (80 kDa), βI (79
kDa), βII (80 kDa), δ (78 kDa) and ɛ (82 kDa) was identified,
whereas expression of PKC ξ (78 kDa) was not identified in the
HRECs.
Proliferation of HRECs under different
conditions
Consistent with the results of a previous study
conducted by our group, the MTT assay demonstrated that moderately
high glucose concentrations and hypoxia conditions (150 μM
CoCl2) significantly induced proliferation of HRECs as
compared with HREC proliferation in the control group. However,
high glucose concentrations alone did not significantly induce cell
proliferation (Fig. 2).
 | Figure 2HREC proliferation and DNA synthesis
following 48 h of treatment at normal glucose (5 mM, Con),
moderately high glucose (15 mM, MG), high glucose (30 mM, HG) and
hypoxia (150 μM CoCl2, HO) conditions. Equal molar
concentrations of mannitol were used for osmotic control; MM, 15 mM
D-mannitol; and HM, 30 mM D-mannitol. (A) Cell proliferation was
assessed using the MTT assay. (B) DNA synthesis was measured as
BrdU incorporation. Data are expressed as the mean ± standard
deviation and the average of four independent experiments.
*P<0.05 vs. control. BrdU, bromodeoxyuridine; HREC,
human retinal endothelial cell. |
Translocation of PKC isoforms under
different conditions
As shown in Fig.
3A, PKC isoform translocation was identified in the moderately
high glucose, high glucose and hypoxia treated groups. PKC δ was
significantly translocated from the cytosol to the membrane in the
moderately high glucose group (P<0.05); whereas PKC α and ɛ were
significantly translocated from the cytosol to the membrane in the
high glucose group. PKC βI and βII were translocated under all
treatment conditions. In the control group, western blot analysis
of the HRECs showed partial translocation to the membrane. Using
confocal fluorescence imaging, the immunofluorescence intensity of
translocated PKC isoforms were enhanced, suggesting an activation
pattern for all of these isoforms (Fig. 3B).
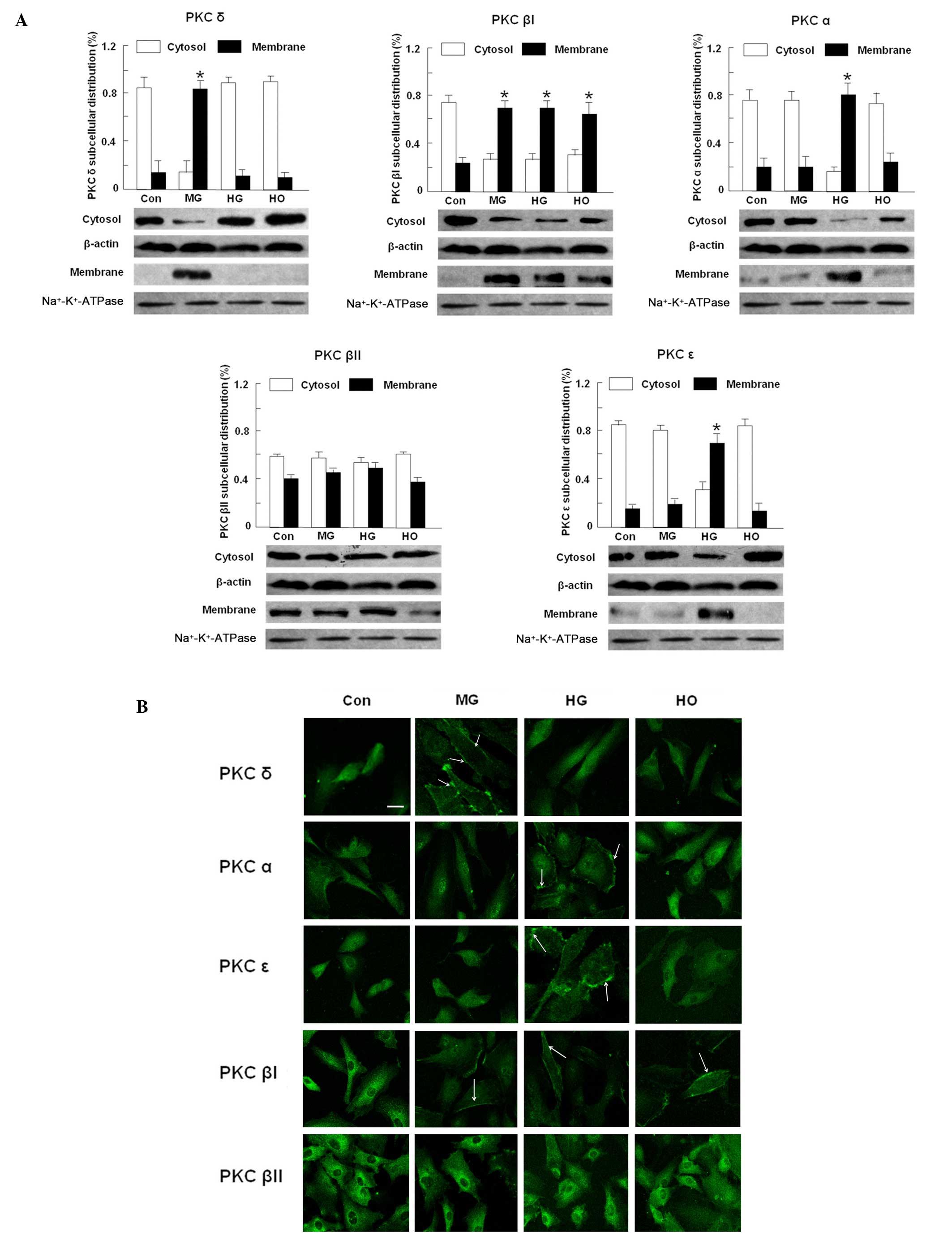 | Figure 3PKC isoform distributions in HRECs
treated at normal glucose (5 mM, Con), moderately high glucose (15
mM, MG), high glucose (30 mM, HG) and hypoxia (150 μM
CoCl2, HO) conditions. (A) Western blot analysis of PKC
isoform distributions in HRECs incubated at different conditions
for 48 h. β-actin was used as the loading control. Data are
expressed as the mean ± standard deviation from three independent
experiments. *P<0.05 vs. control. (B)
Immunofluorescence imaging of PKC isoforms in HRECs treated at
different conditions for 48 h. White arrowheads denote PKC
translocation to the membrane. Scale bar, 20 μm. Original
magnification, ×400. PKC, Protein kinase C; HREC, human retinal
endothelial cell; ATPase, adenosinetriphosphatase. |
Effects of rottlerin and Ro 32-0432 on
cell proliferation
Rottlerin has a high affinity for PKC δ
(IC50 3–6 μM). It has also been reported to inhibit
other isoforms of PKC in addition to PKC δ, but at a concentration
of >30 μM. By contrast, previous studies have demonstrated that
rottlerin inhibited PKC δ at <10 μM (12–15).
The present study identified that rottlerin prevented cell
proliferation triggered at moderately high glucose conditions
(Fig. 4A and B). However,
rottlerin did not elicit any effects on cell proliferation induced
by hypoxic conditions or proliferation under normal glucose
concentrations. Previous studies have found that Ro32-0432, at a
concentration of 200 nM, exerts inhibitory effects on PKC α, β and
ɛ (16,17); however, no significant inhibition
of cell proliferation was observed in the present study (Fig. 4C and D).
 | Figure 4Effects of rottlerin or Ro32-0432 on
HREC proliferation and DNA synthesis. HRECs were treated under
different conditions for 48 h with or without rottlerin (Ro, 10 μM)
or Ro32-0432 (Ro32, 200 nM) in either normal glucose (5 mM, Con),
moderately high glucose (15 mM, MG), high glucose (30 mM, HG) and
hypoxia (150 μM CoCl2, HO) conditions. Equal amounts of
DMSO were used as the vehicle control in all groups except the
control group. (A) Effects of rottlerin on cell proliferation was
measured by the MTT assay. (B) Effect of rottlerin on DNA synthesis
was measured as BrdU incorporation. (C) Effects of Ro32-0432 on
cell proliferation was measured by the MTT assay. (D) Effect of
Ro32-0432 on DNA synthesis was measured as BrdU incorporation. Data
are expressed as the mean ± standard deviation from four
independent experiments. *P<0.05 vs. DMSO and
#P<0.05 vs. MG group. HREC, human retinal endothelial
cell; DMSO, dimethylsulfoxide; BrdU, bromodeoxyuridine. |
Discussion
The present study focused on the effects of high
glucose and hypoxic conditions on cell proliferation, as these
stimuli are considered to be relevant in the initiation and
progression of diabetic retinopathy and other ischemic
retinopathies. In previous studies on diabetes, 25–30 mM is the
most commonly employed glucose concentration in cell models;
however, in the present study a moderately high glucose (15 mM)
concentration was used, which may be closer to the clinic situation
(18), regarding the particularity
of primary HRECs. An incubation time of 48 h was selected in
consideration of the cell growth cycle. It was identified that
these primary HRECs progressed into the exponential phase ~12–24 h
following cell passage. This phase lasted ~48–60 h prior to the
cells entering the silent phase.
Primary cultured HRECs were employed in the present
study in order to reflect the clinic situation. Previous studies
using HRECs, but not other endothelial cells, have obtained similar
results to those of the present study. Premanand et al
(19) failed to identify an
increase in cell proliferation following high glucose (30 mM)
exposure in HRECs, despite observing an increased expression of
vascular endothelial growth factor (VEGF). This finding is
consistent with the results of the present study indicating cell
proliferation under high glucose (30 mM) conditions; however,
Premanand et al did not identify cell proliferation under
moderately high glucose conditions. Notably, this phenomenon may be
due to a particularity of this primary HREC culture. Furthermore,
proliferation of primary cultured human umbilical vascular
endothelial cells was investigated in the present study, and an
opposite result to that of the proliferation of HRECs was obtained:
Moderately high glucose conditions decreased cell proliferation in
a glucose concentration-dependent manner. This was consistent with
a previous study (20).
It was unexpected that moderately high glucose (15
mM) but not high glucose (30 mM) levels significantly triggered
cell proliferation in HRECs. A previous study by our group showed
that VEGF secretion was glucose concentration-dependent, unlike the
proliferation results. This indicated that VEGF may not affect the
proliferation of HRECs, but may be a possible explanation for the
correlation between glucose concentration and proliferation. In the
case of moderately high glucose, which represents clinical
hyperglycemia in untreated patients with diabetes, endothelial
cells may develop certain degrees of tolerance to glucose toxicity.
Therefore, cell proliferation induced by VEGF appeared to be
dominant. By contrast, the high glucose conditions exceeded the
physiologically tolerable range. Glucose toxicity may have been
predominant, so that proliferation was not observed.
Since its seminal discovery by Kishimoto et
al (21), PKC has gained
increasing attention in the biomedical field due to its extensive
role in cell signaling, ion channel regulation, cell proliferation
and differentiation as well as tumorigenesis. The PKC isoforms
display distinct localization and expression in different tissues
and cells. Information regarding expression of PKC isoforms in eye
tissues has been somewhat inconsistent. In the retinal tissue of
rabbits, three isoforms, PKC α, β and γ, were identified (22). In addition, PKC α, βI, βII, δ, ɛ
and ζ were detected in the mouse retina (7). Moreover, PKC α, βI, βII, ɛ and δ were
expressed in rat retina. In human retinal tissues, PKC α, βI, βII,
ɛ, δ, θ, ζ, ι and μ were detected in retinal pigment epithelial
cells (23). However, few studies
have focused on PKC isoforms in HRECs. Therefore, the expression
and translocation of the most common six isoforms of PKC that have
been previously identified in retinal tissues, PKC α, βI, βII, ɛ, δ
and ζ, were examined in the present study. To the best of our
knowledge, the present study was the first to demonstrate the
expression of five PKC isoforms, including PKC α, βI, βII, δ and ɛ,
in primary cultured HRECs, while PKC ζ was not identified. Under
the same experimental conditions, all six isoforms of PKC were
identified in rat brain tissue, which supports the results of the
present study.
The present study aimed to examine the translocation
of PKC isoforms triggered by high glucose, moderately high glucose
and hypoxic conditions in primary cultured HRECs. Cell
fractionation and immunofluorescence imaging demonstrated that
membrane PKC δ levels were markedly increased in the moderately
high glucose group; however, not in the high glucose group. Thus,
it was hypothesized that moderately high glucose and high glucose
levels had discrepant effects on cell proliferation. Previous
studies have reported that PKC δ was widely distributed in various
organs and cell types, and that the excessive activation of PKC δ
may be associated with tumor growth, angiogenesis and neutrophil
adhesion (24–26). In primary cultured HRECs, the
proliferation caused by moderately high glucose levels was
prevented by rottlerin, an inhibitor of PKC δ. Ro32-0432, which is
capable of inhibiting PKC α, βI and ɛ, exhibited only minor
significant effects on cell proliferation. These findings suggest
that PKC δ contributed to cell proliferation under moderately high
glucose conditions.
The pathogenesis of diabetic retinopathy is a
complex process involving multiple factors, among which chronic
activation of PKC has been deemed a prominent factor associated
with vascular alterations, including increased permeability,
contractility, extracellular matrix synthesis, cell growth,
apoptosis and angiogenesis. Among PKC isoforms, previous studies
have reported that PKC α, β, δ and ɛ were activated by
hyperglycemia in retinal tissues (27,28).
Clinical trials have demonstrated positive results for diabetic
non-proliferate retinopathy following the administration of a PKC
βII isoform inhibitor. This supports our findings in which PKC βII
was not associated with the proliferation of primary cultured HRECs
(i.e. proliferative retinopathy).
Although high glucose failed to trigger cell
proliferation, it induced translocation of PKC α and ɛ. In previous
studies, the distinct function and complicated interactions of PKC
isoforms have been reported. For example, decreased PKC α
expression levels resulted in a significant decrease in cell
proliferation in human pigment epithelium cells (29). In addition, PKC δ is able to
regulate PKC α activity (30).
Moreover, treatment of human umbilical vein endothelial cells with
VEGF resulted in the activation of PKC α, but not PKC ɛ, which was
associated with enhanced proliferation and angiogenesis (31). PKC ɛ has been indicated to be
cardioprotective as its activation was required and was sufficient
to induce a preconditioning-like response. By contrast, PKC δ
activation contributed to myocardial damage in ischemia/reperfusion
(32). The same opposing roles of
PKC ɛ and δ have been reported for cerebral ischemia/reperfusion
(33). Therefore, the interactions
among PKC α, ɛ and δ require further investigation in order to
highlight the mechanisms by which high glucose concentrations do
not cause proliferation in HRECs.
The present study revealed that the effects of
hypoxia and moderately high glucose levels on the translocation of
PKC isoforms were different, although they resulted in equal
amounts of cell proliferation. Hypoxia may act through affecting
VEGF expression via the increased binding of the active hypoxia
inducible factor-1α to the hypoxic response element of the VEGF
promoter (34). Although PKC ζ
attenuated hypoxia-induced proliferation of fibroblasts by
regulating MAP kinase phosphatase-1 expression (35), PKC ζ was not expressed in the
present study. Rottlerin, an inhibitor of PKC δ, was capable of
preventing cell proliferation caused by moderately high glucose
levels, but not by hypoxia. Furthermore, PKC δ was not translocated
under hypoxic conditions. These results suggest that PKC δ was not
associated with cell proliferation caused by hypoxia. This
conclusion is further supported by the fact that Ro32-0432, which
inhibits several PKCs other than PKC δ, did not affect cell
proliferation at medium high glucose levels.
Acknowledgements
This study was supported by the Project of National
Ministry of Science and Technology (no. 2011ZX11101) and Guangdong
Province (no. 2007A032702001).
References
|
1
|
Lu M, Kuroki M, Amano S, et al: Advanced
glycation end products increase retinal vascular endothelial growth
factor expression. J Clin Invest. 101:1219–1224. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kern TS and Engerman RL: Capillary lesions
develop in retina rather than cerebral cortex in diabetes and
experimental galactosemia. Arch Ophthalmol. 114:306–310. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Agardh CD, Agardh E, Zhang H and Ostenson
CG: Altered endothelial/pericyte ratio in Goto-Kakizaki rat retina.
J Diabetes Complications. 11:158–162. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Crawford TN, Alfaro DV 3rd, Kerrison JB
and Jablon EP: Diabetic retinopathy and angiogenesis. Curr Diabetes
Rev. 5:8–13. 2009. View Article : Google Scholar
|
|
5
|
Das Evcimen N and King GL: The role of
protein kinase C activation and the vascular complications of
diabetes. Pharmacol Res. 55:498–510. 2007.PubMed/NCBI
|
|
6
|
Steinberg SF: Structural basis of protein
kinase C isoform function. Physiol Rev. 88:1341–1378. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pedro G and George LK: Activation of
protein kinase C isoforms and its impact on diabetic complications.
Circ Res. 106:1319–1331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JY, Takahara N, Gabriele A, et al:
Induction of endothelin-1 expression by glucose: an effect of
protein kinase C activation. Diabetes. 49:1239–1248. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao R, Zhu BH, Tang SB, et al:
Scutellarein inhibits hypoxia- and moderately-high glucose-induced
proliferation and VEGF expression in human retinal endothelial
cells. Acta Pharmacol Sin. 29:707–712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lai P, Li T, Yang J, et al: Upregulation
of stromal cell-derived factor 1 (SDF-1) expression in
microvasculature endothelial cells in retinal ischemia-reperfusion
injury. Graefes Arch Clin Exp Ophthalmol. 246:1707–1713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li X, Hahn CN, Parsons M, et al: Role of
protein kinase C zeta in thrombin-induced endothelial permeability
changes: inhibition by angiopoietin-1. Blood. 104:1716–1724. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sakai H, Yamamoto M, Chiba Y, et al: Some
different effect of PKC inhibitors on the acetylcholine, and
endothelin-1-induced contractions of rat bronchial smooth muscle.
Eur J Pharmacol. 618:58–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wong C and Jin ZG: Protein kinase
C-dependent protein kinase D activation modulates ERK signal
pathway and endothelial cell proliferation by vascular endothelial
growth factor. J Biol Chem. 280:33262–33269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Griger Z, Páyer E, Kovács I, et al:
Protein kinase C-β and -δ isoenzymes promote arachidonic acid
production and proliferation of MonoMac-6 cells. J Mol Med.
85:1031–1042. 2007.
|
|
15
|
Kim JH, Kim JH, Jun HO, et al: Inhibition
of protein kinase C attenuates blood-retinal barrier breakdown in
diabetic retinopathy. Am J Pathol. 176:1517–1524. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilkinson SE, Parker PJ and Nixon JS:
Isoenzyme specificity of bisindolylmaleimides, selective inhibitors
of protein kinase C. Biochem J. 294:335–337. 1993.PubMed/NCBI
|
|
17
|
Ding M, Huang C, Lu Y, et al: Involvement
of protein kinase C in crystalline silica-induced activation of the
MAP kinase and AP-1 pathway. Am J Physiol Lung Cell Mol Physiol.
290:L291–L297. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eitel I, Hintze S, de Waha S, et al:
Prognostic impact of hyperglycemia in nondiabetic and diabetic
patients with ST-elevation myocardial infarction insights from
contrast-enhanced magnetic resonance imaging. Circulation:
Cardiovascular Imaging. 5:708–718. 2012.
|
|
19
|
Premanand C, Rema M, Sameer MZ, et al:
Effect of curcumin on proliferation of human retinal endothelial
cells under in vitro conditions. Invest Ophthalmol Vis Sci.
47:2179–2184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rojas S, Rojas R, Lamperti L, et al:
Hyperglycaemia inhibits thymidine incorporation and cell growth via
protein kinase C, mitogen-activated protein kinases and nitric
oxide in human umbilical vein endothelium. Exp Physiol. 88:209–219.
2003. View Article : Google Scholar
|
|
21
|
Kishimoto A, Takai Y and Nishizuka Y:
Activation of glycogen phosphorylase kinase by a calcium-activated,
cyclic nucleotide-independent protein kinase system. J Biochem.
82:1167–1172. 1977.PubMed/NCBI
|
|
22
|
Osborne NN, Barnett NL, Morris NJ, et al:
The occurrence of three isoenzymes of protein kinase C (alpha, beta
and gamma) in retinas of different species. Brain Res. 570:161–166.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu KM, Ma P, Ge J, et al: Expression of
protein kinase C isoforms in cultured human retinal pigment
epithelial cells. Graefes Arch Clin Exp Ophthalmol. 245:993–999.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Keshamouni VG, Mattingly RR and Reddy KB:
Mechanism of 17-β-estradiol-induced Erk1/2 activation in breast
cancer cells. A role for HER2 and PKCδ. J Biol Chem.
277:22558–22565. 2002.
|
|
25
|
Abbas T, White D, Hui L, et al: Inhibition
of human p53 basal transcription by down-regulation of protein
kinase Cδ. J Biol Chem. 279:9970–9977. 2004.PubMed/NCBI
|
|
26
|
Steinberg SF: Distinctive activation
mechanisms and functions for protein kinase C delta. Biochem J.
384:449–459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Inoguchi T, Battan R, Handler E, et al:
Preferential elevation of protein kinase C isoform beta II and
diacylglycerol levels in the aorta and heart of diabetic rats:
differential reversibility to glycemic control by islet cell
transplantation. Proc Natl Acad Sci USA. 89:11059–11063. 1992.
View Article : Google Scholar
|
|
28
|
Idris I, Gray S and Donnelly R: Protein
kinase C activation: isozyme-specific effects on metabolism and
cardiovascular complications in diabetes. Diabetologia. 44:659–673.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao Q, Tan J, Ma P, et al: PKC alpha
affects cell cycle progression and proliferation in human RPE cells
through the downregulation of p27kip1. Mol Vis. 15:2683–2695.
2009.PubMed/NCBI
|
|
30
|
Murakami M, Horowitz A, Tang S, et al:
Protein kinase C (PKC) delta regulates PKC alpha activity in a
Syndecan-4-dependent manner. J Biol Chem. 277:20367–20371. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wellner M, Maasch C, Kupprion C, et al:
The proliferative effect of vascular endothelial growth factor
requires protein kinase C-alpha and protein kinase C-zeta.
Arterioscler Thromb Vasc Biol. 19:178–185. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bright R and Mochly-Rosen D: The role of
protein kinase C in cerebral ischemic and reperfusion injury.
Stroke. 36:2781–2790. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Murriel CL and Mochly-Rosen D: Opposing
roles of delta and epsilon PKC in cardiac ischemia and reperfusion:
targeting the apoptotic machinery. Arch Biochem Biophys.
420:246–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aiello LP, Northrup JM, Keyt BA, et al:
Hypoxic regulation of vascular endothelial growth factor in retinal
cells. Arch Ophthalmol. 113:1538–1544. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Short MD, Fox SM, Lam CF, et al: Protein
kinase Cζ attenuates hypoxia-induced proliferation of fibroblasts
by regulating MAP kinase phosphatase-1 expression. Mol Biol Cell.
17:1995–2008. 2006.
|