Introduction
Osteogenesis imperfecta (OI), also known as brittle
bone disease, characterized by increased bone fragility and low
bone mass, is a rare inherited connective tissue disease (1,2),
mainly with autosomal dominant inheritance. The incidence of OI is
~1:10,000 and the capital clinical manifestations of OI are
multiplicative osteopsathyrosis, blue sclera, dentinogenesis
imperfecta and mild audition (3).
The traditional classification is based on clinical and
radiographic criteria, and the mode of inheritance findings include
type I (MIM #166200), II (MIM #166210), III (MIM #259420) and IV
(MIM #166220) (4,5). Their phenotypic presentation varies
from mild to lethal, of which type I is the mildest with the
highest incidence rate.
More than 90% patients with OI types I–IV have
autosomal dominant mutations in one of the two genes that encode
the α chains of type I collagen. Type I collagen, the main
component of bone matrix, consists of two pro-α1 chains and one
pro-α2 chain, which are encoded by COL1A1 (MIM #120150;
accession# for mRNA Z74615.1) and COL1A2 (MIM #120160;
accession# for mRNA Z74616.1) genes, respectively. COL1A1 is
fixed on chromosome 17q21.3–17q22 and COL1A2 on chromosome
7q22.1, their total lengths are 18 and 38 kb, respectively,
containing ~50 exons. Furthermore, each α chain consists of 1,014
amino acids forming a triple helical domain encoded by 43 exons.
The folding of the three chains, with limited space, with triple
helical domain restricts every third residue to a Gly, generating a
repeating (Gly-X-Y)n sequence pattern. Once Gly in triple helix
domains is substituted with other amino acids, the structure of
type I collagen is destroyed. Gly is significant role in the
formation of the triple helical domain, these sites are also the
molecular basis of numerous diseases.
A number of gene mutations associated with
COL1A1 and COL1A2 have been reported resulting in OI.
To date, hundreds of mutations referring to COL1A1 and
COL1A2 have been submitted to the University of Leicester’s
(Leicester, UK) database (http://www.le.ac.uk/genetics/collagen/) highlighting
mutations of the collagen gene (6). A Chinese patient with OI type I
disease, who possessed a novel single base substitution mutation
(c.3263G>A) in the COL1A1 gene was observed. This
identification is likely to add to the the mutation database of
collagen genes and provide more abundant materials for clinical
diagnosis of heritable diseases and aid in the development of gene
therapy and molecular diagnosis in the antepartum period.
Materials and methods
Case presentation and analysis
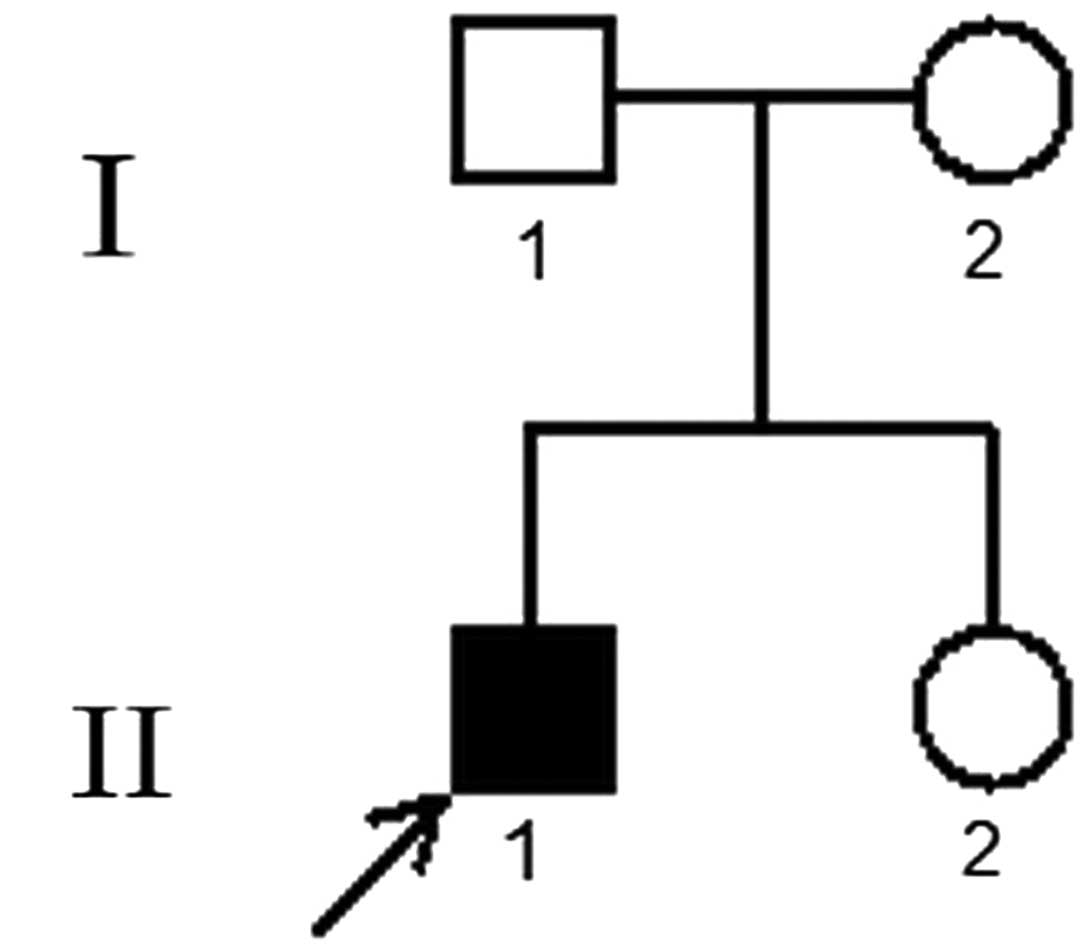
A 15-year-old Chinese male (Fig. 1) came to the Jinling Hospital
(Nanjing, China) for genetic counseling of constitutional bone
disease, which was characterized by blue sclera and mild
abnormalities with the teeth. The patient was 171 cm tall and
weighed 54 kg, with normal vision, audition and intelligence.
According to the patient, he had previously suffered six fractures
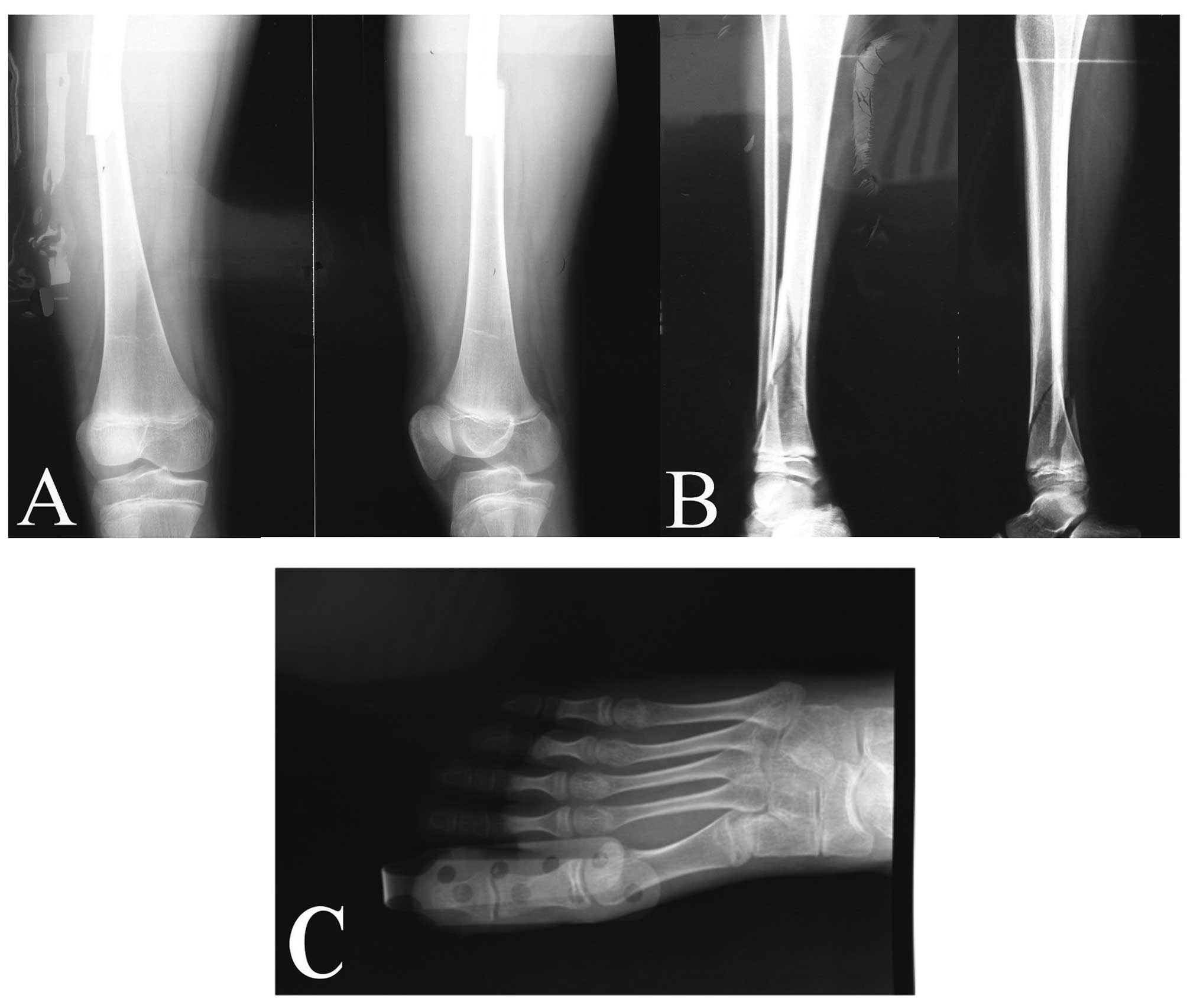
in the four limbs. Among these, foot dislocation was the latest
fracture in 2010, and the most serious was a comminuted fracture in
the right tibiofibula in 2006 (Fig.
2). The patient’s parents and sister were all healthy. In
addition, 250 healthy adults were used as controls.
DNA sequencing
A peripheral blood sample (5 ml) was obtained from
the patient, the patient’s parents and sister, and 250 normal
adults, respectively. All instructions were performed by the
Institutional Review Board and the Committee on Ethics of Research
Involving Human Subjects of Nanjing University Medical Center,
Nanjing, China (7). The Wizard™
Genomic DNA Purification kit (Promega Corporation, Madison, WI,
USA) was used to extract leucocyte genomic DNA, which was stored at
-20°C, according to the manufacturer’s instructions. The primers
were designed according to ~50 exons of COL1A1 and
COL1A2 (8),
respectively, and polymerase chain reaction (PCR) was
completed under the following conditions: 95°C for 5 min followed
by 35 cycles of 95°C for 30 sec, 56°C for 30 sec, and 72°C for 60
sec (8). PCR products were
sequenced directly using an ABI Prism 3700 automated sequencer
(Applied Biosystems, Foster City, CA, USA). The sequencing results
were compared with the NCBI Reference Sequence (NM_000088).
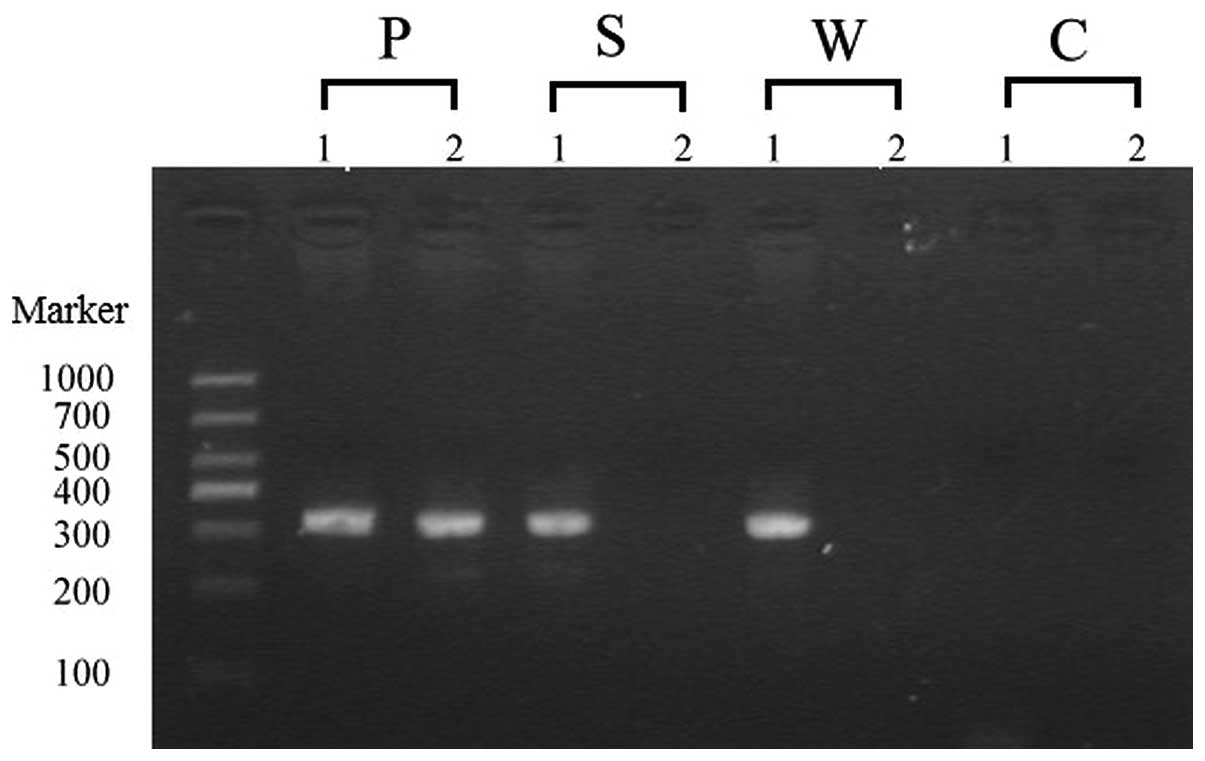
PCR-sequence specific primer (SSP)
experiments
Allele-specific primer extension was used to confirm
the heterozygous state. The wild-type gene sequence was amplified
with primer A (5′-TGTCGCTTTTTC TCTTGTAGGG-3′) plus primer C
(5′-CTCGCATAATCGCTCTTTGTGTA-3′) and the mutation sequence was
amplified with primer B (5′-TGTCGCTTTTTCTCTTGTAGGA-3′) plus primer
C. It was expected that the products of wild-type and mutation-type
sequences generated a fragment of 315 bp. The PCR cycling products
were visualized using a UV transilluminator (WD-9403B; Beijing
Liuyi Instrument Factory, Beijing, China) in a 2.0% agarose gel
following electrophoresis and staining with ethidium bromide.
Results
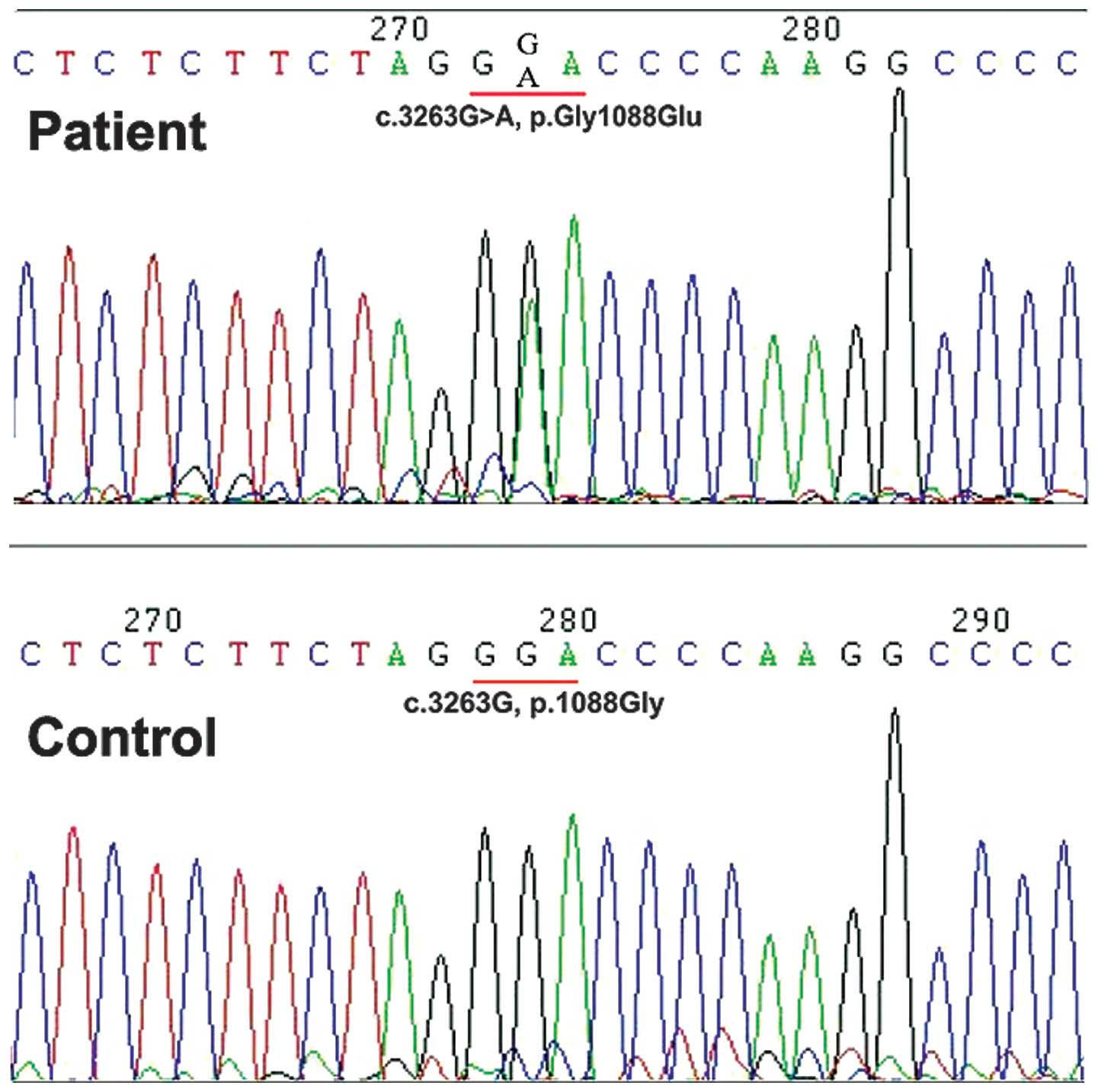
A single base substitute mutation, c.3263G>A (p.
Gly1088Glu), was identified in the sequence analysis of the
COL1A1 gene. The number of this mutation site was based on
the cDNA sequence found in GenBank for human mRNA pre-pro-αI (I)
(GenBank accession no. Z74615.1), using the A of ATG translation
initiation start site as nucleotide +1 (9). This mutation (c.3263G>A) was shown
in the spectrum of the group (Fig.
3), the same mutation was not identified in the patient’s
parents or sister, or the 250 unrelated controls. The novelty of
this mutation was identified, which was not defined in the Type I
Collagen Mutation Database (http://www.le.ac.uk/genetics/collagen/), or the Human
Gene Mutation Database (http://www.uwcm.ac.uk/uwcm/mg/hgmd().html). This
mutation was further confirmed by PCR-SSP in the schematic diagram
(Fig. 4). There also existed a
single-nucleotide polymorphism in exon 33 (c.2298T>C) in this
sporadic case, no other mutations were identified.
Discussion
There are numerous mutations in the prolyl
3-hydroxylation complex, including CRTAP (MIM #605497), LEPRE1 (MIM
#610339) and PPIB (MIM #123841), and the non-collagen related
genes, FKBP10 (MIM #607063), PLOD2 (MIM #601865), SERPINF1 (MIM
#172860), SERPINH1 (MIM #600943) and SP7 (MIM #606633). These lead
to defective bone development and abnormal collagen helix formation
associated with a clinical spectrum of recessive OI (10). In addition, type I–IV of OI were
almost all triggered by mutations in COL1A1 and
COL1A2. Type I collagen, encoded by COL1A1 and
COL1A2, consists of two pro-α1 chains and one pro-α2 chain.
Each α chain includes an N- and C-terminal, and a core triple
helical domain of the Gly-X-Y triplet repeat unit, which is highly
conserved. The C-terminal is used for recognition and assembly of
type I collagen (11,12), and is cleaved until the triple
helical domain is assembled into type I collagen. The N-terminal
domain is also cleaved. According to the protein sequences in NCBI,
with the reference sequence NM_000088.3, there are 1442 amino acids
in the collagen pro-α1 (I) chain. The N-terminal domain consists of
approximately the first 200 amino acids and the C-terminal domain
consists of almost all the last 200 amino acids. (13). The remaining amino acids form the
triple helix domain. Substitution mutations in the first 200 amino
acids, occur only in the N-terminal domain, thus are nonlethal and
exhibit a variable outcome. The triple helix domain is not
considered as a domain purely, but rather as being composed of
subdomains, each having its own specific function. The sizes of
these subdomains remain unknown; however, the biochemical features
of the substitution amino acids are linked to the role of the
regions where the mutations occur.
The patient with OI type I had a guanine to adenine
transition at nucleotide c.3263G>A in exon 45 that may result in
a Gly to Glu substitution, which corresponds to the glycine
position of Gly-X-Y repeat units within the triple helix domain of
the collage protein (14). This
variant was not identified in the patient’s parents or sister, or
250 controls. As shown in an alignment of COL1A1 orthologs
from six species of M. musculus, R. norvegicus, C.
familiaris, D. rerio, M. mulatta, and H.
sapiens, the original mutation position in COL1A1 is
highly conserved across a number of species (Fig. 5). That suggests a substitution for
Gly across the triple helix domain may induce different types of OI
regardless of the substitution. However, the collagen gene
mutations were not located in already existing hot spots.
Maurizia et al (15) reported a proband with disease of a
lethal form of OI, in which an 18-month-old female succumbed to
respiratory failure. The study showed that a G to C conversion at
nucleotide 3263 of pro-α1 (I) (c.3263G>C), which may change Gly
to Ala in the triple helix domain. The mutation site was the same
as that reported in the current study, however resulted in
substitution with a different amino acid. The triple helix chain
bearing these point mutations may exhibit secretion difficulties
and the extensive mutable trimers may obstruct the secretion of
normal chains. Furthermore, the chains containing one or two
mutations exhibited poor thermal stability. The different
substituted amino acids were hypothesized to result in a number of
abnormalities, including collagen recognition, anomalous assembly
and functional abnormality. To the best of our knowledge, this is
the first this is the first study to demonstrate that disparate
substitutions of amino acids at the same position may result in
entirely different phenotypes from mild to lethal. It provided
evidence for further establishment of phenotype-genotype
associations, and it also suggested the foundations for the
determination of the molecular mechanism underlying the development
of OI.
In conclusion, a novel Gly substitution mutation
(c.3263.G>A) in exon 45 of COL1A1 gene resulting in OI
type I was identified in a sporadic patient. The detailed molecular
and clinical features are likely to be useful for extending the
evidence for genetic and phenotypic heterogeneity and exploring the
phenotype-genotype correlations in OI.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 30901652), the Key
foundation of Jiangsu Science and Technology Bureau (grant no.
BM2013058) and the Foundation of Jiangsu province (grant no.
BK2011660). The authors would like to thank all members of the
family for their cooperation in the study.
References
|
1
|
Byers PH: Disorders of collagen
biosynthesis and structure. The Metabolic and Molecular Basis of
Inherited Disease. Scriver CR, Beaudet AL, Sly WS and Valle D: 7th
edition. McGraw-Hill; New York, NY: pp. 4029–4077. 1995
|
|
2
|
Byers PH, Wallis GA and Willing MC:
Osteogenesis imperfecta: translation of mutation to phenotype. J
Med Genet. 28:433–442. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pollitt R, McMahon R, Nunn J, et al:
Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with
osteogenesis imperfecta type I–IV. Hum Mutat. 27:7162006.
|
|
4
|
Morello R, Bertin TK, Chen Y, et al: CRTAP
is required for prolyl 3-hydroxylation and mutations cause
recessive osteogenesis imperfecta. Cell. 127:291–304. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Dijk FS, Nesbitt IM, Zwikstra EH, et
al: PPIB mutations cause severe osteogenesis imperfecta. Am J Hum
Genet. 85:521–527. 2009.PubMed/NCBI
|
|
6
|
Marini JC, Forlino A, Cabral WA, et al:
Consortium for osteogenesis imperfecta mutations in the helical
domain of type I collagen: regions rich in lethal mutations align
with collagen binding sites for integrins and proteoglycans. Hum
Mutat. 28:209–221. 2007. View Article : Google Scholar
|
|
7
|
Xia XY, Cui YX, Huang YF, et al: A novel
RNA-splicing mutation in COL1A1 gene causing osteogenesis
imperfecta type I in a Chinese family. Clin Chim Acta. 398:148–151.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Körkkö J, Ala-Kokko L, De Paepe A,
Nuytinck L, Earley J and Prockop DJ: Analysis of the COL1A1 and
COL1A2 genes by PCR amplification and scanning by
conformation-sensitive gel electrophoresis identifies only COL1A1
mutations in 15 patients with osteogenesis imperfecta type I:
identification of common sequences of null-allele mutations. Am J
Hum Genet. 62:98–110. 1998.
|
|
9
|
Lee KS, Song HR, Cho TJ, et al: Mutational
spectrum of type I collagen genes in Korean patients with
osteogenesis imperfecta. Hum Mutat. 27:5992006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Forlino A, Cabral WA, Barnes AM and Marini
JC: New perspectives on osteogenesis imperfecta. Nat Rev
Endocrinol. 7:540–557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boot-Handford RP and Tuckwell DS:
Fibrillar collagen: the key to vertebrate evolution? A tale of
molecular incest. Bioessays. 25:142–151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Exposito JY, Cluzel C, Garrone R and
Lethias C: Evolution of collagens. Anat Rec. 268:302–316. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stover DA and Verrelli BC: Comparative
vertebrate evolutionary analyses of type I collagen: potential of
COL1a1 gene structure and intron variation for common bone-related
diseases. Mol Biol Evol. 28:533–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weng YC, Sonni A, Labelle-Dumais C, et al:
COL4A1 mutations in patients with sporadic late-onset intracerebral
hemorrhage. Ann Neurol. 71:470–477. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valli M, Sangalli A, Rossi A, et al:
Osteogenesis imperfecta and type I collagen mutations A lethal
variant carsed by a Gly910 - Ala substitution in the α1(I) chain.
Eur J Biochem. 211:415–419. 1993.PubMed/NCBI
|