1. Introduction
Pim was initially identified by cloning the
retroviral integration sites in murine Moloney leukaemia virus
(MMLV)-induced lymphomas (1). Pim
is a member of the family of oncoproteins that exhibit
serine/threonine kinase activity (1). Furthermore, all Pim proteins were
confirmed to contain an active site, termed the adenosine
triphosphate (ATP) anchor. Pim genes represent a family of
proto-oncogenes that encode three different kinases (Pim-1, -2 and
-3) belonging to the Ca2+/calmodulin-dependent protein
kinase group, essential in the regulation of signal transduction
cascades (2). Pim kinases are
evolutionarily conserved, exhibiting a high degree of homology in
sequence and structure (3). Pim
kinases are normally constitutively active and are broadly
expressed in haematopoietic, vascular smooth muscle and epithelial
cell lineages, as well as in embryonic stem cells. Thus, they are
essential for the normal growth and maturation of these cells.
MMLV proviral insertion in the 3′-untranslated
region (UTR) of Pim-1 led to an increase in the stability of
Pim-1 mRNA and Pim-1 protein expression, resulting in
tumourigenesis of the T-cell lymphoma (4). Moreover, Pim-1 can prevent apoptosis
and promote cell proliferation, effects which are considered
important for malignant transformation (5). Furthermore, elevated Pim-1 expression
levels have been observed in human haematopoietic malignancies as
well as in solid tumours (6–8).
Under physiological circumstances, the body tightly
controls the expression of the genes responsible for regulation of
cell growth, apoptosis and the cell cycle. However, in a number of
pathological conditions, dysregulation of these genes can lead to
cellular dysplasia and induce malignant transformation of cells.
Pim-1 is an oncogene that is important in the regulation of
cell growth. The crystal structure of Pim-1 reveals the absence of
an identified regulatory domain, which indicates that it does not
depend on post-transcriptional modifications for activation and
thus is constitutively active. Therefore, regulation of Pim-1
kinase activity largely depends on its protein expression level
rather than its phosphorylation level (9). Notably, Pim-1 expression is
not only transcriptionally mediated by a number of molecules but is
also transiently induced by microRNAs and hormones at the
post-transcriptional level. The mechanisms of cell gene expression
regulation are diverse (10). In
the present review, the regulation of Pim-1 expression and a
number of its inhibitors are discussed, providing theoretical
guidance for the development of molecular targeting therapies and
drug treatments for Pim-1-associated diseases.
2. Structure and biological functions of
Pim-1
In humans, the Pim-1 oncogene, ~5 kb in
length, is located on the short arm of chromosome 6p21.1-p21.31.
The mRNA transcript for Pim-1 is encoded by six exons with
large 5′- and 3′UTRs containing a G/C-rich region and five copies
of AUUA destabilising motifs (2).
Pim-1 can generate two isoforms (34 and 44 kD) due to
alternative translation initiation points at an upstream CUG codon.
The shorter form localises to the cytoplasm and the nucleus,
whereas the longer form localises to the plasma membrane; however,
the two proteins retain their serine/threonine kinase activity. The
oncogenic activity of Pim kinases is mediated by multiple cellular
substrates (6). Pim-1 kinase
adopts a two-lobed kinase fold structure with a deep cleft between
the N- and C-terminal lobes connected via the hinge region
(residues 121–126). The N-terminal lobe is composed primarily of
β-sheets, whereas the C-terminal lobe is comprised of α-helices.
The hinge region has specific residues identified as ATP-binding
sites. Moreover, the ATP-binding pocket in Pim-1 is always open,
indicating that Pim-1 kinase constitutively resides in an active
conformation (9,11,12).
The Pim-1 oncogene is frequently
overexpressed in a range of haematological malignancies and several
solid tumours. Its activity supports tumour cell growth and
survival in vitro and in vivo through phosphorylation
of a large number of common substrates, including several cell
cycle regulators and apoptosis mediators. Pim-1 kinase can
phosphorylate Cdc25A (G1/S), Cdc25C (G2/M), p21CIP1/WAF1
and p27, all of which are involved in the regulation of the cell
cycle (7,13–16).
It is also able to inactivate Bad protein by phosphorylating
ser112, impeding the process of apoptosis and supporting
tumour cell growth and survival. Additionally, it interacts with
the nuclear mitotic apparatus protein to promote mitosis (2). Thus, Pim-1 may alter various
biological functions to accelerate oncogenesis by inhibiting
apoptosis, enhancing cell proliferation and promoting cell
differentiation.
3. Regulators of Pim-1 expression
Regulation at the transcriptional
level
Similar to other protein kinases, Pim-1 expression
is known to be regulated mainly by the rate of transcription.
Binding of a wide range of growth factors, hormones and cytokines,
such as interleukins, epidermal growth factor, prolactin,
granulocyte colony-stimulating factor and granulocyte-macrophage
colony-stimulating factor, to target surface-specific receptors
activates the Janus kinase/signal transducer and activator of
transcription (JAK/STAT) signal transduction pathway, which is
essential for regulating Pim-1 gene expression. Janus
kinases subsequently phosphorylate the cytoplasmic receptor
tyrosine kinase domain, thus generating recruitment sites for STATs
containing the SH2 domain. The activation of STATs phosphorylated
by JAK leads to their dimerisation and nuclear translocation. In
the nucleus, STAT3 and STAT5 directly bind to the Pim-1
promoter at the ISFR/GAS sequence, thus upregulating the
transcription of Pim-1. In addition, Pim-1 itself is able to
negatively regulate the JAK/STAT pathway by binding to a group of
negative regulators, termed suppressor of cytokine signalling
proteins (2). In
BCR/ABL-expressing oncoprotein cells, Pim-1 is markedly upregulated
following activation of BCR-ABL tyrosine kinase by activation of
STAT5 (17). Moreover, nuclear
factor-κB can rapidly induce Pim-1 expression following
tumour necrosis factor-α stimuli in a STAT5-mediated manner
(18). Thus, multiple signalling
pathways have STATs at their core, forming a complex network to
co-regulate Pim-1 expression at the transcriptional level
(Fig. 1).
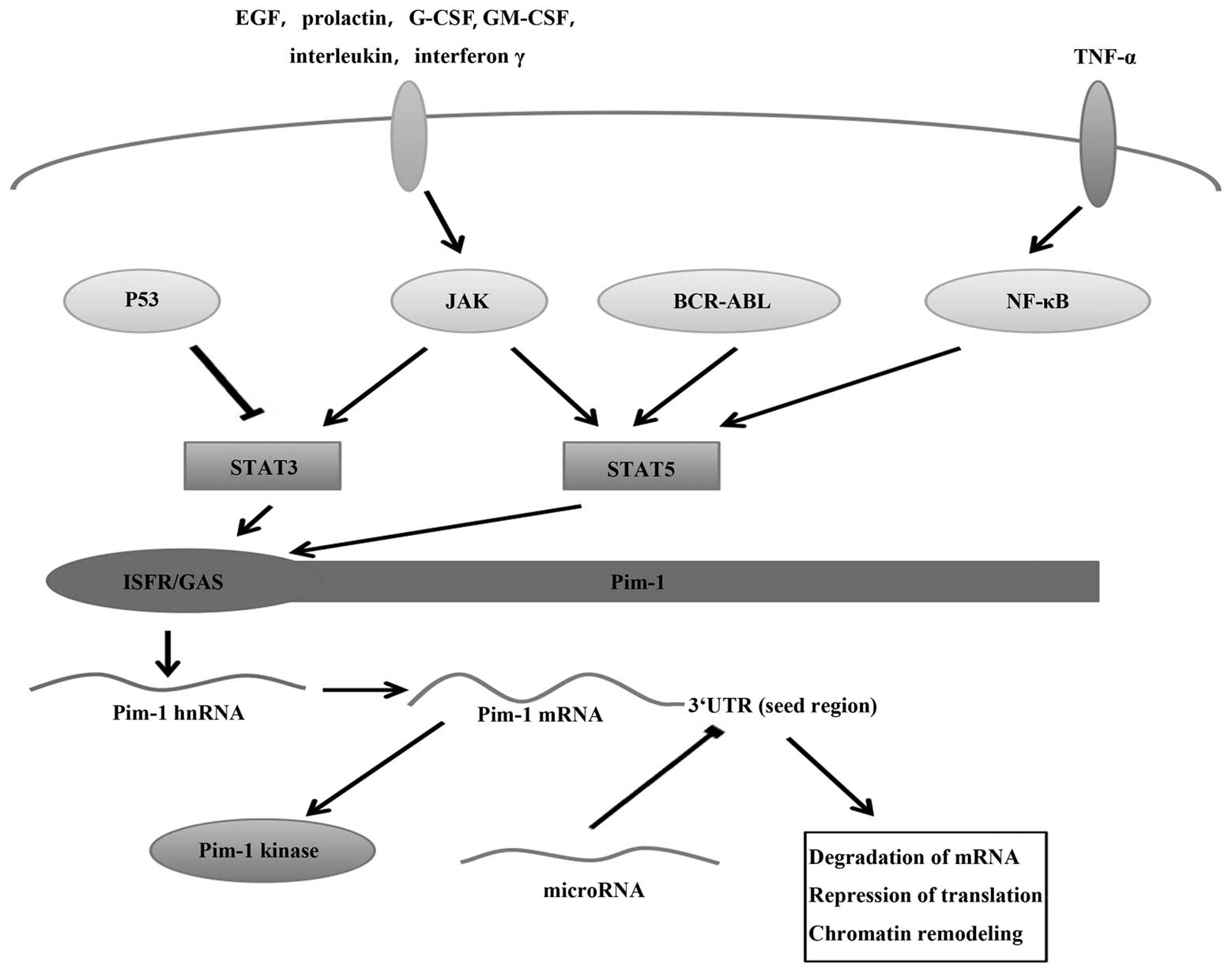 | Figure 1Regulation of Pim-1 at the
transcriptional level via the JAK/STAT signalling pathway and at
the post-transcriptional level via miRNA. Receptor stimulation by
hormones, cytokines and growth factors, such as interleukins,
interferon γ, EGF, prolactin, G-CSF and GM-CSF, activate STAT by
JAKs, BCR-ABL and NF-κB. p53 can inhibit STAT3. STAT3 and STAT5
upregulate the transcription of Pim-1 by binding to the Pim1
promoter at the ISFR/GAS-sequence, since STAT proteins serve as
transcription factors for Pim genes following engagement of their
cognate ligands. miRNAs can bind to the seed region in the Pim-1
3′UTR, inhibiting Pim-1 expression to result in mRNA
destabilisation and translational inhibition. JAK, Janus kinase;
STAT, signal transducer and activator of transcription; miRNA,
microRNA; EGF, epidermal growth factor; G-CSF, granulocyte
colony-stimulating factor; GM-CSF, granulocyte-monocyte
colony-stiumulating factor; 3′UTR, 3′-untranslated region; TNF,
tumour necrosis factor; hnRNA, heterogenous nuclear RNA. |
Regulation via knockdown of Pim-1
expression using microRNA
MicroRNAs (miRNAs) are a family of small noncoding
RNA molecules, ~22 nucleotides (nt) in length, which
post-transcriptionally silence target gene expression to act as
either oncogenes or tumour-suppressor genes (19–21).
Several miRNAs have been identified to be associated with
Pim-1-related tumour initiation. Downregulation of Pim-1 by miRNAs,
which may contribute to the differential expression of Pim-1 in
tumours versus normal cells, regulates the cell cycle and
apoptosis. Analysis of the structure of human Pim-1 mRNA indicates
that the 3′UTR of Pim-1, which is generally evolutionarily
conserved, harbours multiple binding sites for miRNAs (22). Additionally, the ‘seed region’
covering nucleotides 2–8 of the mature miRNA strand is essential
for interacting with the target miRNA to destabilise it and inhibit
its translation (23,24) (Fig.
1).
miRNA-16 acts as an important tumour suppressor by
regulating pro-oncogene expression (25). Various types of cancer cells,
including chronic lymphocytic leukaemia (CLL) and prostate cancer
cells, have been shown to reduce miRNA-16 levels (26,27).
Fms-like tyrosine kinase 3 (FLT3), is expressed in a large number
of acute myeloid leukaemia (AML) cases, and is activated by
internal tandem duplication (ITD) mutations, which aberrantly
activate its downstream signalling to contribute to cell
proliferation and inhibit apoptosis (28). This may explain why AML patients
with the FLT3/ITD mutant phenotype have a poor clinical prognosis
(28–31). Notably, in FLT3/ITD-expressing
cells, Pim-1 is upregulated and is involved in FLT3-mediated cell
survival (32,33), whereas miRNA-16 is downregulated.
Furthermore, based on bioinformatics analysis, miRNA-16 may target
the 3′UTR of human Pim-1 (22,34).
Using quantitative polymerase chain reaction and immunoblotting,
Pim-1 mRNA and protein levels were confirmed to be markedly
decreased upon miRNA-16 mimic transfection in FLT3/ITD-expressing
cells. Therefore, miRNA-16 appears to bind to the 3′UTR putative
target site of Pim-1 and directly inhibit its expression at the
post-transcriptional level, thus slowing down the growth of
FLT3/ITD-expressing cells (34).
However, enforced miRNA-16 expression could not completely deplete
Pim-1 expression in FLT3/ITD-expressing cells, suggesting
continuous induction of Pim-1 by other FLT3/ITD-mediated signalling
molecules (34). Activation of
FLT3 can also activate numerous signal transduction pathways,
including STAT5 phosphorylation (35–37),
for the upregulation of Pim-1 (38). Thus, Pim-1 may be a predominant
downstream target of multiple signalling pathways activated by
FLT3. Notably, miRNA-16 expression is comparably high in the K562
myeloid leukaemia cell line and the LS174T colon cancer cell line,
but miRNA-16 did not affect Pim-1 expression in either cell
line (22). In conclusion,
FLT3/ITD and Pim-1 are important in the transformation of leukaemia
cells. This evidence supports the hypothesis that FLT3 should be
routinely analysed to guide therapy and estimate prognosis in AML
patients, and may be used as a novel target for chemotherapeutics
to treat patients with leukaemia. Moreover, miRNA-16 is an
important component of the FLT3/ITD signalling pathway, and it may
serve as a clinically useful biomarker and permit the development
of novel drugs.
miRNA-33 has two isoforms, miRNA-33a and miRNA-33b,
involved in the regulation of gene transcription associated with
cholesterol biosynthesis and uptake (39), but their relevance with regard to
tumours has rarely been explored. Notably, in a variety of cancer
cell lines, miRNA-33a expression has been revealed to be generally
low, with Pim-1 expression relatively high. miRNA-33a significantly
reduces Pim-1 expression in K562 and LS174T cells via specifically
binding to the seed region of Pim-1. The repression effects were
essentially abrogated in the presence of a mutated seed region
(22). However, miRNA-33a was not
shown to affect Pim-1 expression in FLT3/ITD expressing cells, thus
the regulation is cell-specific (34). In MM.1S multiple myeloma cells,
Pim-1 is also a direct target of miRNA-33b, participating in the
apoptosis induced by miRNA-33b (40). However, miRNA-33b is not considered
to be the primary miRNA to regulate Pim-1 in K562 and LS174T cells,
although levels of miR-33b are observed to be low in the two cell
lines (22).
Ibrahim et al (41) preclinically validated a
polyethylenimine (PEI)-mediated method of unmodified miRNA-33a
delivery in a mouse model of colon carcinoma through in
vitro and in vivo experiments. This method can
efficiently strengthen the repression of Pim-1 expression in
vivo in tumour cells, and its antitumour effect is similar to
that of Pim-1 knockdown using small interfering (si)RNA/PEI.
Moreover, the inhibition effect of the modified miRNA-33a mimic
resembles that of siRNAs fully complementary to the miRNA-33a
target site, but is more efficient than the unmodified mimic
(22). Thus, this approach using
miRNAs for cancer therapy is expected to become an efficient and
biocompatible strategy for targeting Pim-1.
miRNA-1 expression is highest in various types of
muscle cell, including spindle-shaped and vascular smooth muscle
cells, mediating cell proliferation and differentiation in cardiac
and skeletal muscle cells (42,43).
Notably, Pim-1 is able to promote proliferation of cultured smooth
muscle cells (SMCs) and neointimal hyperplasia in vitro
(44). Chen et al (45) demonstrated that miRNA-1 is a
downstream effector of myocardin and inhibits the proliferation of
SMCs mediated by myocardin. Furthermore, miRNA-1 may mediate the
repression of Pim-1 expression at the translational level to
inhibit vascular SMC proliferation. In conclusion, miRNA-1 is able
to inhibit SMC proliferation and may be a novel target site for
vascular smooth muscle proliferative diseases.
Diabetic cardiomyopathy was characterised as having
reduced Pim-1 levels in the early stages of the disease, along with
reduced upstream activators, p-STAT3 and p-Akt (46). Notably, in a model of diabetic
cardiomyopathy, miRNA-1 was upregulated from an early phase and was
positively correlated with the progression of diabetic
cardiomyopathy, indicating that miRNA-1 can negatively modulate
Pim-1 to mediate the further progression of cardiomyopathy
(47). Katare et al
(47) performed in vitro
cell experiments demonstrating that miRNA-1 not only inhibited
Pim-1 directly but also via its upstream modulator, Akt. Moreover,
anti-miRNA-1 could restore Pim-1 expression and the proliferative
activity of cardiac progenitor cells. Thus, an imbalance between
negative and positive regulators leads to the repression of Pim-1
with advancing cardiomyopathy. This provides insight into novel
strategies for gene therapy for this disease.
Although miRNA-1 levels in other tissues are lower
than in the muscle (48),
comparison of genomic positions of mouse tumour susceptibility loci
with those of mouse miRNA genes revealed that the flanking region
of miRNA-1 has six substitutions affecting susceptibility to lung
cancer (49). It has previously
been reported that in human primary lung cancer tissues and almost
all lung cancer cell lines, miRNA-1 expression is comparably low,
whereas Pim-1 expression is significantly upregulated (12,50,51).
A study has revealed that miRNA-1 binding to the 3′UTR of Pim-1
negatively affects regulation of the antitumour effect of Pim-1
(50). Thus, the inhibitory
mechanism of Pim-1 expression mediated by miRNA-1 exists not only
in muscle cells, but also in tumour cells. As miRNA-1 has diverse
roles in various diseases, due to the wide distribution in levels
of miRNA-1 expression, further studies into its regulation of
tumour-associated angiogenesis are warranted.
Several studies have determined that Pim-1 kinase is
important in the survival of BCR/ABL+ cells (52). miRNA-328 expression is
downregulated, whereas Pim-1 protein is markedly upregulated in
BCR/ABL+ cells. One study has demonstrated that
miRNA-328 interacting with the 3′UTR of Pim-1 could inhibit Pim-1
expression, blocking cell proliferation and growth (53).
miRNA-210 is inducible by hypoxia and appears to be
a hypoxia-inducible factor target gene (54,55).
Huang et al (56)
identified Pim-1 as an miRNA-210 target gene through the
miRNP-IP approach followed by cloning the 3′UTR of Pim-1 to
perform reporter assays. However, luciferase activity was repressed
by 15% in the Pim-1 construct compared with co-transfection
with a control plasmid. In addition, the inhibitory effect of
miRNA-210 on tumour growth initiation was not rescued by expressing
the Pim-1 coding sequence without the 3′UTR. These
observations suggest that Pim-1 may be a weak miRNA-210
target gene, although Pim-1 is enriched by microarray
analysis.
In conclusion, the expression of various miRNAs
depends on the pathological cell type and multiple stimuli. miRNA
can simultaneously regulate multiple target genes and Pim-1 is
regulated by multiple miRNAs. Notably, different primary miRNAs
regulate Pim-1 expression in different tissues and cells. Thus, in
tumour cells with Pim-1 overexpression, identifying the miRNAs
important in regulating Pim-1 expression requires further research.
Moreover, the 3′UTR of Pim-1 harbours numerous other miRNA binding
sites, that may be important in the development of various
diseases.
Regulation of Pim-1 expression by
hormones
Previous studies have shown that one of the major
risk factors of breast cancer is cumulative oestrogen exposure
(57). Oestrogen receptor α (ER-α)
inhibits Forkhead box protein M1 expression, which is involved in
the occurrence and development of breast cancer induced by
oestrogen (58). It has been
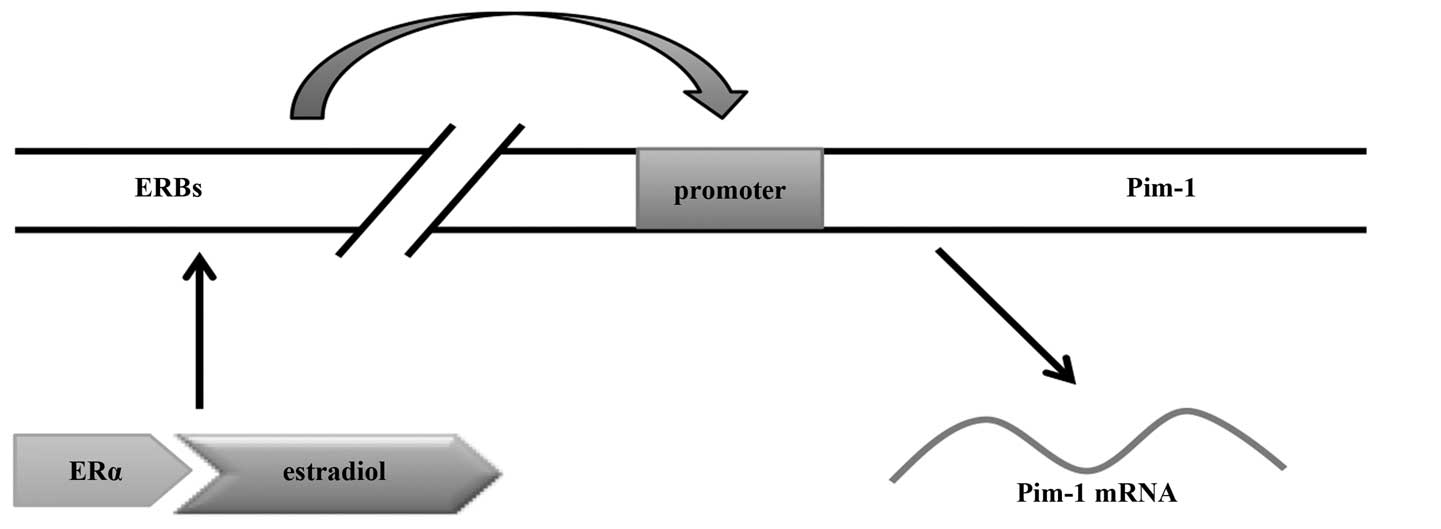
recently reported that there are four binding sites for oestradiol
(E2)-loaded ER-α far upstream of the Pim-1 promoter, and
these ER-α-binding regions (ERBs) may function as
oestrogen-regulated enhancers for Pim-1 (59). Oestradiol rapidly triggers loading
of ER-α to the ERBs, then ERBs interact with each other via
chromatin loop formation, resulting in Pim-1 expression.
Thus, Pim-1 is a direct ER-α target in breast cancer cells and
oestradiol positively regulates its expression in an ER-α-mediated
manner (Fig. 2). Furthermore,
Pim-1 overexpression induced by oestradiol has been
determined to phosphorylate and thereby inhibit the expression of
cell cycle inhibitors, hindering apoptosis, promoting cell cycle
progression and increasing invasiveness of breast cancer tumours.
Collectively, these results add a novel potential mechanism by
which oestradiol is able to promote breast cancer cell
proliferation (59).
Dehydroepiandrosterone (DHEA) is an abundantly
produced steroid hormone, known to improve pulmonary arterial
hypertension (PAH) through its vasodilator properties and reverse
vascular remodelling (60). Paulin
et al (61) investigated
pulmonary artery smooth muscle cells (PASMCs) in PAH, and
demonstrated a significant decrease in the p-STAT3/STAT3 ratio and
the nuclear translocation of p-STAT3 following treatment of the
PAH-PASMCs with DHEA. Similarly, DHEA also decreases Pim-1
mRNA and protein levels in PAH-PASMCs. Since Pim-1 is the
STAT3 downstream target implicated in PAH, the results suggest that
DHEA can downregulate Pim-1 expression via decreasing the quantity
of phosphorylated STAT3 for activation in PAH-PASMCs.
Pim-1 overexpression in prostate cancer cells
has been associated with tumourigenesis (62). Preclinical data regarding Vitamin
D3 (calcitriol) have revealed antiproliferative and
apoptosis-inducing effects resulting in significant antitumour
activities in prostate cancer cells (62,63).
Since calcitriol treatment can result in hypercalcaemia, the dose
that can be administered to patients is less than that
theoretically required for antitumour activity. Consequently,
Okamoto et al (64)
synthesised and tested inecalcitol, a novel and unique analogue of
vitamin D3 that is potent but less calcaemic. Pim-1 expression was
shown to be decreased in a dose-dependent manner following
treatment of LNCaP prostate cancer cells and LNCaP xenografts with
inecalcitol or calcitriol, respectively. In addition, inecalcitol
was more potent than calcitriol in downregulating the levels of
Pim-1 mRNA and protein.
It is reported that Pim-1 overexpression can
downregulate the androgen receptor (AR)-mediated signalling that
inhibits cell proliferation and induces dedifferentiation by AR
phosphorylation (65). Notably,
Pim-1 is closely associated with hormone refractory prostate
cancers (65). Therefore,
administration of inecalcitol may have a positive impact on the
therapy of androgen-dependent prostate cancer.
Conversely, Maier et al (66) identified that Pim-1 kinase may not
be a calcitriol target gene in HaCaT keratinocytes. However, Pim-1
can interact with the vitamin D3 receptor (VDR) DNA-binding domain,
participating in signal transduction of calcitriol. Further
research into the mechanism involved in the interaction of Pim-1
and calcitriol is required.
Androgens are the key male hormones involved in the
development of the prostate gland. Androgens can promote the
development of androgen-dependent prostate cancer mediated through
the androgen receptor, which is a key hormone correlated with
prostate cancer, and androgen deprivation therapy is a common
treatment for prostate cancer patients (67,68).
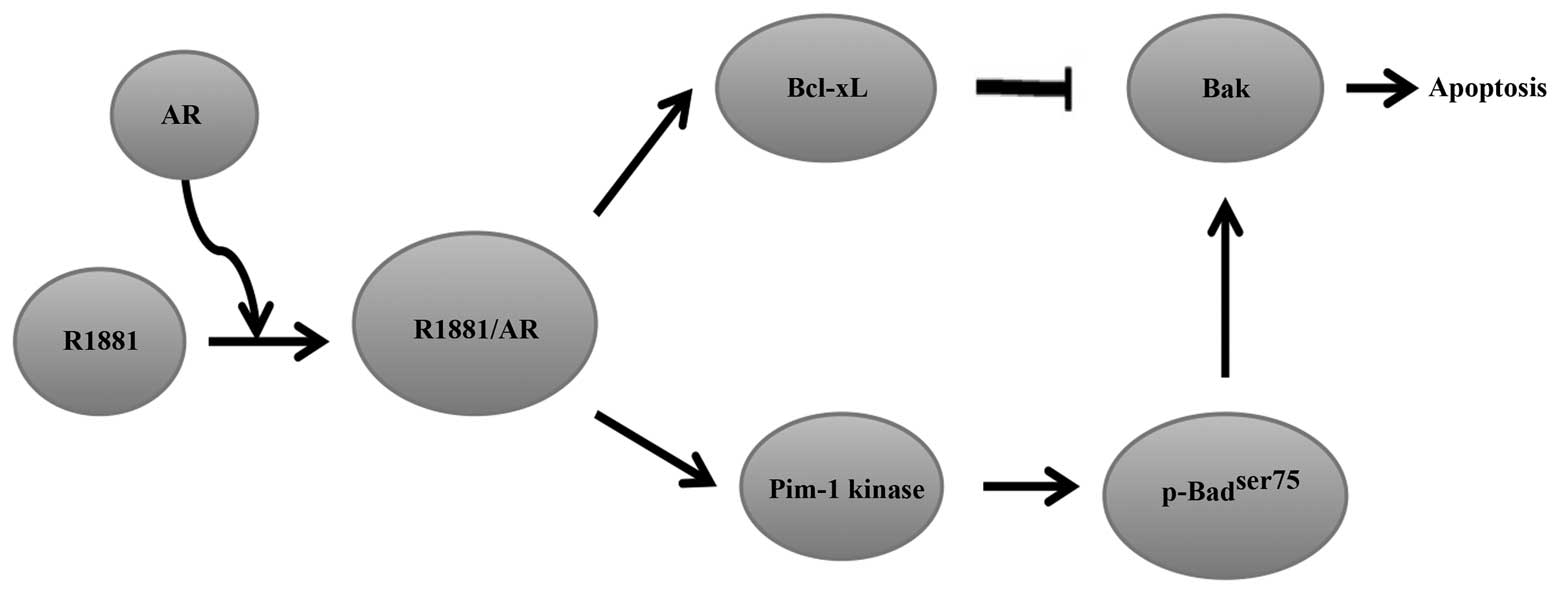
The phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 can
induce apoptosis in serum-deprived LNCaP prostate cancer cells. The
apoptosis-inducing effect is significantly neutralised by androgen
methyltrienolone, R1881, resulting in cell survival and
proliferation (69). Moreover, the
prosurvival effects of R1881 are linked to Bcl-xL overexpression
(70). Kumar et al
(71) demonstrated that activation
of AR by R1881 induced an increase in Bcl-xL expression, which
contributed to sequestering the pro-apoptotic protein Bak, thereby
preventing its activation and the accompanying prosurvival effects.
In addition, the authors confirmed that the pro-survival effect of
Bcl-xL requires an increase in the stability of protein kinase
Pim-1. Furthermore, the results indicated that the increase of
Pim-1 kinase activity and stability correlated with an increase in
the half-life of Pim-1 by R1881 induction rather than an increase
in the transcription rate. Notably, R1881-induction was not caused
by an increase in the quantity of Pim-1 protein. The enhanced
activity of Pim-1 kinase prevented full activation of Bad via
phosphorylation of Bad at ser75 and offset
dephosphorylation of Bad by LY294002, and enhanced Bcl-xL to exert
its anti-apoptotic activity through the sequestration of Bak
(Fig. 3) (71). In conclusion, these results have
improved the understanding of the molecular mechanism of
tumourigenesis of prostate cancer to provide novel insights for the
treatment of androgen-dependent prostate cancer.
The emerging identification of the importance of
hormone imbalance in the development of human tumours has increased
interest in the development of hormone-associated drugs. As more is
revealed concerning the hormone-associated mechanisms of
Pim-1 expression, more will be understood about the
association between hormones and tumour development. Targeting
Pim-1 may offer a strategy for improved treatment of
hormone-dependent tumours.
Regulation of Pim-1 expression by
PI3K-like kinases
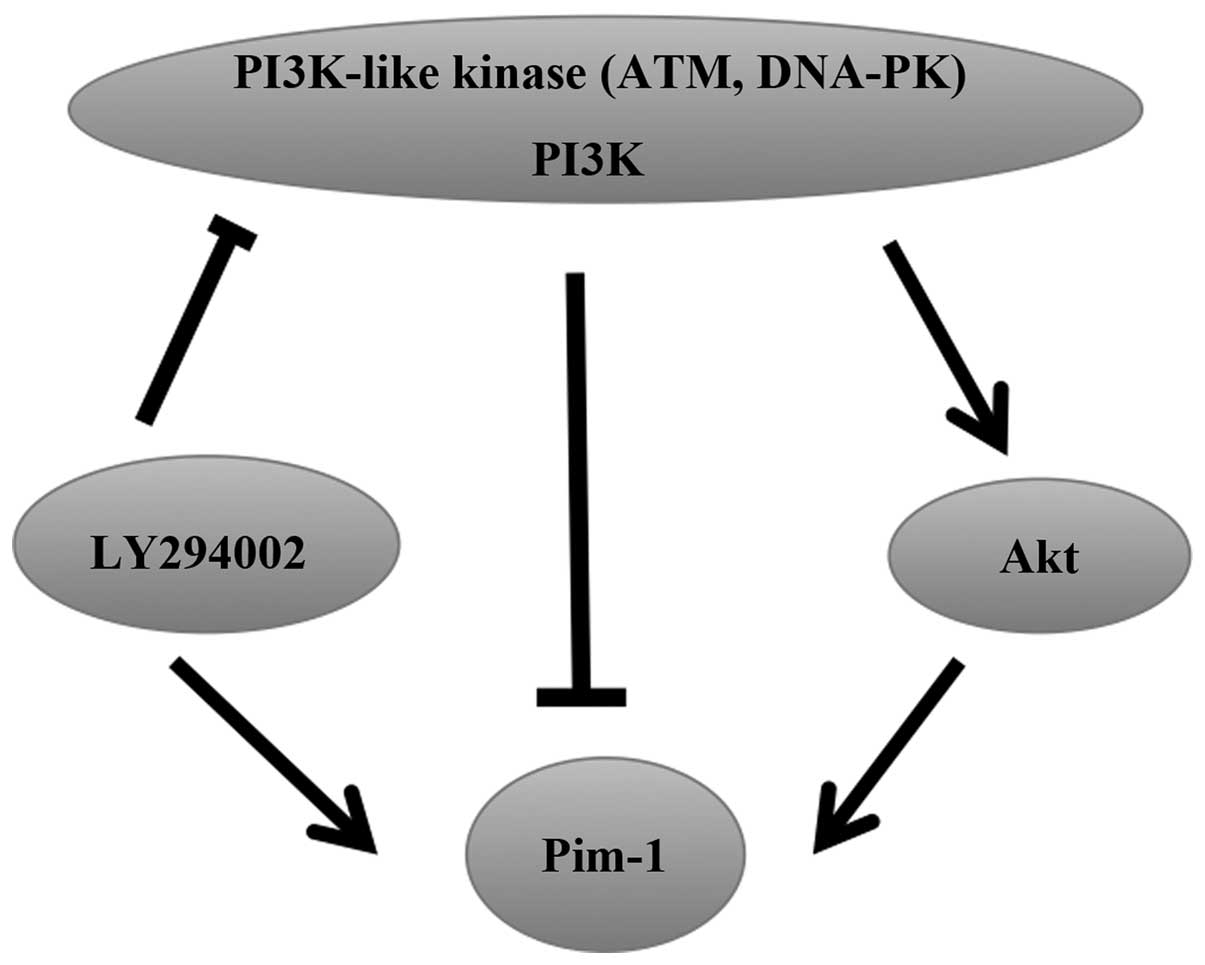
DNA-dependent protein kinase complex (DNA-PK) and
Ataxia-Telangiectasia Mutated (ATM) are members of the PI3K-like
kinase family. Akt is a downstream effector of PI3K and LY294002 is
an inhibitor of PI3K-like kinases. The expression of Pim-1, Pim-2
and Pim-3 mRNA is rapidly increased following treatment of
endothelial cells (ECs) with LY294002, but there is no effect on
the stability of the mRNA, indicating that LY294002 can regulate
the activity of the promoters of Pim to induce the
upregulation of Pim expression (72). Akt overexpression has been reported
to increase Pim-1 expression in neonatal rat cardiomyocytes
(73). Similarly, Pim-1 is a
crucial downstream target of Akt in ECs, and Akt can increase
Pim-1 expression, but does not affect Pim-2 and
Pim-3 expression. In addition, DNA-PK and ATM can decrease
Pim-1 expression in physiological conditions (72). Thus, PI3K-like kinases have dual
effects on the regulation of Pim expression (Fig. 4).
Regulation of Pim-1 expression by
cytokines
The vascular endothelial growth factor
(VEGF)-A/Flk-1 signalling pathway also increases the Pim-1
expression level. Zippo et al (74) identified Pim-1 as a target
gene of Flk-1, which is induced during the process of angiogenesis.
Furthermore, in angiogenesis of human umbilical cord vein
endothelial cells, VEGF-A can induce Pim-1 expression
mediated by Flk-1, although this induction is poor with
Pim-1 expression levels only marginally increased.
Consistent with this finding, this mechanism also exists in
vivo in ECs during angiogenesis of the ovary (74). However, platelet-derived growth
factor bb (PDGFbb), but not VEGF-A165, can transcriptionally
stimulate Pim-1 expression in vascular smooth muscle cells
(VSMC), mostly attributable to the activation of JAK/STAT, but also
to an additional pathway involving protein kinase C (PKC) and the
mitogen-activated protein kinase Mek1/2, leading to the expression
of the Pim-1 kinase and proliferation of VSMCs (75,76).
Moreover, it has been recently reported that interleukin (IL)-6
stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell lines
and that the increase in IL-6-stimulated Pim-1 may be partially
STAT3-independent (77).
4. Pim-1 inhibitors
ATP mimetic inhibitors
ATP mimetic inhibitors bound to Pim-1 are sandwiched
between hydrophobic residues from the glycine-rich loop, the
C-terminal domain of Pim-1 kinase and the hinge region. The
presence of proline at position 123 prevents the molecules from
forming the second hydrogen bond to the hinge, thereby only one
hydrogen bond between the ligand and the hinge is observed
(78). These inhibitors comprise
the broad-spectrum kinase inhibitor staurosporine and its analogue
K252, bisindoylmaleinimides and the related PKC inhibitor LY333531,
as well as a number of extremely potent organometallic inhibitors
(18). Medical research into this
class of inhibitors has thus far been limited.
ATP competitive inhibitors
ATP competitive inhibitors do not interact with the
hinge region by forming classical hydrogen bonds so can therefore
be considered as ATP competitive inhibitors and not ATP mimetic
inhibitors. SGI-1776, SMI-4a, LY294002, quercetagetin,
1,10-dihydropyrrolo[2,3-α]carbazole-3-carbaldehyde (DHPCC-9) and
more recently, pyrrolo[2,3-g]indazoles have been idenfied as ATP
competitive inhibitors(79). A
number of these compounds are in phase I clinical trials (80,81).
SGI-1776 is an imidazo[1,2-β]pyridazine small
molecule inhibitor. Certain studies have reported that multiple
prostate cancer, leukaemia, lymphoma and multiple myeloma cell
lines treated with SGI-1776 exhibited a significant reduction in
the phosphorylation levels of traditional Pim-1 substrate proteins,
histone H3, c-Myc and Bad, interfering with proliferation and
viability (40,82–84).
These data suggest that SGI-1776 can induce apoptosis by inhibiting
Pim-1 function and producing a cytotoxic effect. SGI-1776 also has
a relatively specific effect against certain paediatric cancers
in vitro and in vivo with selected activated kinases
at SGI-1776 concentrations, but it is more effective against AML
(81). Furthermore, cells exposed
to increasing doses of SGI-1776 arrested in a dose-dependent manner
in the G1 cell cycle, inhibiting the natural progression to S
phase. This was followed by apoptosis, as determined by measuring
the caspase-3 activity, correlating with the downregulation of
p21waf1 and Bad phosphorylation (82). Conversely, unlike in replicating
cells, phosphorylation of traditional Pim-1 kinase targets was
unaffected by SGI-1776 in CLL, indicating an alternative mechanism
to induce apoptosis (85). In
addition, treatment of the DU-145 prostate cancer cell line and the
MiaPaCa2 pancreatic cancer cell line with SGI-1776 resulted in a
significant reduction in p-STAT3Tyr705 expression
without affecting STAT3 expression and STAT5 phosphorylation,
suggesting specificity for p-STAT3Tyr705. The inhibitory
effect of SGI-1776 on STAT3Tyr705 phosphorylation is
primarily mediated by Pim-3 in DU-145 cells (86). Siu et al (87) subsequently determined that the
upregulation of MIG6 induced by SGI-1776 involved Pim-1 and that
MIG6 may be a target gene of Pim-1. Recently, SGI-1776 was revealed
to recover the sensitivity to doxorubicin in p-glycoprotein
(ABCB1)-overexpressing cells (88). It was further identified that
SGI-1776 could decrease cell surface expression of ABCB1 and the
breast cancer resistance protein ABCG2 (which are substrates of
Pim-1) and drug transport by Pim-1-dependent and Pim-1-independent
mechanisms (89). Notably,
SGI-1776 can resensitise chemoresistant cells to taxane-based
therapies by inhibiting multidrug resistance activity and inducing
apoptosis (82). Combination with
cytarabine can increase the efficacy of Ara-C, significantly
decreasing the viability of AML cell lines (90). Therefore, SGI-1776 can retard cell
growth in several human haematological malignancies and solid
tumours in vitro. However, phase I clinical trials have not
been successful due to the cardiotoxicity of the drug. As a result,
this prompted the development of antitumour drugs with more
antitumour effects and fewer side effects based on the structure of
this compound (91).
Quercetagetin is a type of flavonol that is also an
inhibitor identified to have a moderately potent antitumour
activity. Holder et al (92) demonstrated that quercetagetin
reduces Pim-1 activity in intact RWPE2 prostate cancer cells in a
dose-dependent manner to cause cell growth arrest, but it exhibited
no effect on AKT kinase. The reducing effect of quercetagetin was
similar to that of knockdown by siRNAs. Furthermore, the inhibitory
ability of quercetagetin on cell growth was proportional to the
quantity of Pim-1 protein in the target cells, particularly at
lower drug concentrations. In addition, vascular SMCs markedly
increased Pim-1 expression upon stimulation with PDGFbb. However,
quercetagetin was able to effectively block this effect, inhibiting
vascular SMC proliferation induced by PDGFbb (76). Treatment of nasopharyngeal
carcinoma cell lines with quercetagetin has been demonstrated to
significantly decrease cell viability, colony formation rate and
migration ability via inhibition of Pim-1 overexpression (93).
The pyrrolo[2,3-α]carbazole has been identified as a
novel scaffold on which to design potent Pim kinase inhibitors. In
addition, several pyrrolo[2,3-α]carbazole derivatives have been
identified that target Pim-1 and Pim-3 with greater selectivity
than Pim-2 under in vitro conditions. The structure of this
inhibitor, which has a non-ATP mimetic binding mode with no
hydrogen bonds formed with the kinase hinge region, is the reason
for the high selectivity of these derivatives for Pim kinases and
its modest but significant selectivity for Pim-3 (94). DHPCC-9 is a potent cellular
inhibitor of these derivatives, which can enter the cells and
completely abrogate the anti-apoptotic effects of Pim-1 to reduce
the viability of cytokine-deprived myeloid cells, whilst not
exhibiting general cytotoxicity at the micromolar concentrations
used. DHPCC-9 reduces all family Pim kinase activities via
inhibition of the phosphorylation of their downstream substrate,
Bad, whilst not reducing their endogenous expression. Moreover,
DHPCC-9 removed the promigratory advantage of Pim by decreasing the
motility of adherent cancer cells in a dose-dependent manner
towards Pim downstream targets, such as nuclear factor of activated
T-cells, cytoplasmic 1. The reduction of cell migration in
vitro by Pim-specific siRNA interference is lower than that
caused by DHPCC-9, which may be due to the longer half-life and
superior cell penetrance of DHPCC-9. Thus, DHPCC-9 is not only an
efficient tool to research the physiological effects of the Pim
family kinases, but also an attractive compound for
chemotherapeutic drug development to prevent tumour metastasis or
angiogenesis by inhibiting the invasiveness of cells overexpressing
Pim (95).
Recently, researchers performed a virtual screening
campaign that led to the identification of a series of
2-aminothiazole derivatives classified as possible allosteric
inhibitors of Pim. This is a novel mechanism of inhibition that is
noncompetitive with respect to ATP and the peptide substrate.
Administering a combination of ATP-competitive and
ATP-noncompetitive compounds highlighted a synergistic effect on
the inhibition of cell proliferation in more highly metastatic cell
lines, where all Pim-1 inhibitors analysed showed synergism with
the known anti-cancer agent, paclitaxel. These results further
establish these derivatives as promising adjuvant agents for the
treatment of cancer in which Pim-1 is associated with
chemotherapeutic resistance (96).
As awareness of the functions of Pim family kinases
in tumourigenesis and the identification of an increasing number of
novel and selective Pim kinase inhibitors has increased, the
investigation and development of small molecule inhibitors
targeting Pim kinases has attracted greater attention. Inhibitors
of Pim kinases have been developed and synthesised; however, only
certain inhibitors have been validated to have antitumour activity
through cell-based assays or animal models, and only a small
proportion of those can effectively inhibit the three members of
the Pim family (82,85,92,94).
Thus, it is important to design and synthesise suitable
chemotherapeutic drugs based on the three-dimensional structure of
Pim kinase to inhibit its activity. Combinations of Pim inhibitors
together with other chemotherapeutics may lead to more efficient
therapeutic approaches.
5. Conclusion
Considering the gradually increasing prevalence of
tumours and their high mortality rates, tumour prevention and
treatment is a key area of medical research worldwide. Alternative
targeting therapies, a novel direction of tumour treatment, are
becoming more important as tumour incidence increases anually. The
study and development of novel chemotherapeutic drugs is confronted
with great opportunities and challenges. With extensive research in
the field of gene therapy, the mechanisms by which Pim-1 expression
is regulated are being increasingly emphasised. It is known that
Pim-1 kinase, identified as an oncogene, is constitutively active
and aberrantly expressed in a number of types of tumours.
Additionally, Pim kinases are involved in the development of
resistance against radiation therapy or chemotherapy. Functional
interference with Pim-1 kinase has been recently reported to impair
the growth and survival of cancer cells. As the structure and
biological functions of Pim-1 are further recognised and regulators
of Pim-1 expression identified, it is clear that Pim-1 has an
impact on the cell cycle and apoptosis under physiological and
pathological conditions. The inhibition of Pim-1 kinase expression
and its activity is significant for the design and development of
chemotherapeutics to treat cancer. Thus, in this review, novel
strategies for tumour therapy from regulators and inhibitors of
Pim-1 are discussed. Pim-1 expression is mainly regulated at the
transcriptional level. However, a number of biomolecules can also
mediate its expression at other levels.
MicroRNAs have emerged as a novel class of noncoding
genes involved in regulating cell proliferation, differentiation
and viability by knockdown of their target genes. Different cell
types have different miRNA expression profiles and different
stimuli can also activate the expression of different miRNAs.
Identifying these stimuli and the regulatory miRNAs requires
further study, which may contribute to the understanding of the
complete signal pathways involving Pim-1. Moreover, an miRNA can
simultaneously regulate multiple target genes, and Pim-1 is
regulated by multiple miRNAs. It was hypothesised that all miRNAs
could directly or indirectly regulate Pim-1 expression and
subsequently regulate cell viability and survival. In the present
study, certain miRNAs, which bind to the seed region of the Pim-1
3′UTR to lead to mRNA destabilisation, have been comprehensively
reviewed according to the present literature. These miRNAs do not
all exhibit key roles in the same types of cells and tissues. These
miRNAs have been identified as tumour suppressor genes, as they can
induce tumour cell apoptosis and inhibit tumour cell proliferation
by repressing Pim-1 expression. They may be able to act as
biomarkers in the research of alternative therapies targeting
Pim-1. However, it remains unclear which miRNA are significantly
involved in other Pim-1 overexpression tumour cells and whether the
expression of miRNAs are tissue-specific. Moreover, the 3′UTR of
Pim-1 harbours other miRNA binding sites, as determined by
computational predictions (22).
The association of miRNA regulation of Pim-1 expression and the
development of relevant diseases remains unexamined.
Hormone imbalance has long been known to be relevant
in the development of human tumours. The association between
hormones and tumours has been further recognised in recent years,
and more hormones have been revealed to be associated with Pim-1
kinase. These hormones can regulate Pim-1 via different pathways to
influence tumour progression and certain biological
characteristics. For example, oestrogen induces Pim-1 expression
via ERBs in the promoter region, whereas DHEA decreases Pim-1 mRNA
and protein levels via p-STAT3. With increasing recognition of the
functional importance of hormones targeting Pim-1 in tumourigenesis
and identification of the relevant molecular mechanisms, improved
choices in the treatment of hormone-dependent tumours can be
developed. However, the effect of hormones is systemic and diverse,
therefore pharmacological and clinical trials are required before a
chemotherapeutic that targets hormones could be adopted.
A number of studies have paid increased attention to
cytokines that regulate the impact of Pim-1 kinase on the tumour
microenvironment (74–77,97).
In this review, certain ATP mimetic inhibitors and ATP competitive
inhibitors are discussed. Notably, a novel mechanism of inhibition
has been recently shown to be noncompetitive with respect to ATP
and the peptide substrate. Using the mechanism, inhibitors can
effectively repress the activity of Pim kinases, promoting tumour
cell apoptosis. Thus it is of importance for the treatment of
cancer to design and develop chemotherapeutic drugs targeting Pim-1
using inhibitor scaffolds. Due to functional redundancy,
simultaneous targeting of all family of Pim kinases can be
advantageous in tumour therapy. However, only a few selective Pim
kinase inhibitors, developed through experiments in vivo and
in vitro, have exhibited antitumour activity, mainly through
targeting Pim-1 and Pim-2. Moreover, certain inhibitors have not
passed phase I clinical trials due to cytotoxicity. Thus, continued
investigation into the crystal structure of all Pim kinases is
required to identify further scaffolds inhibiting kinase activity.
These results also suggest the need to consider the structure of
the compound to develop antitumour drugs with more potential
antitumour effects and fewer side effects. These inhibitors are
still in the preliminary stages of development.
Although certain regulatory mechanisms of Pim remain
unknown, the development of therapeutic agents targeting
therapeutic genes in tumours in which Pim-1 is aberrantly expressed
may become a novel research focus. Despite the numerous questions
and obstacles that remain, it is hoped that the combined
application of inhibitors of Pim-1 expression and Pim-1-specific
inhibitors together with other anticancer strategies may provide
novel and efficient therapies for cancer patients.
Acknowledgements
The authors would like to acknowledge grant support
from the National Science Foundation of China (NSFC) (grant nos.
30973476 and 812727), the Shanghai Pujiang Programme (grant no.
KW201028464), Fudan University ‘985 Project’ Phase III Cancer
Research Projects II (grant no. 985III-YFX0102) and the Shanghai
Committee of Science and Technology (grant no. 12DZ2260100).
References
|
1
|
Cuypers HT, Selten G, Quint W, et al:
Murine leukemia virus-induced T-cell lymphomagenesis: integration
of proviruses in a distinct chromosomal region. Cell. 37:141–150.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bachmann M and Möröy T: The
serine/threonine kinase Pim-1. Int J Biochem Cell Biol. 37:726–730.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mikkers H, Nawijn M, Allen J, et al: Mice
deficient for all PIM kinases display reduced body size and
impaired responses to hematopoietic growth factors. Mol Cell Biol.
24:6104–6115. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Selten G, Cuypers HT, Boelens W, et al:
The primary structure of the putative oncogene pim-1 shows
extensive homology with protein kinases. Cell. 46:603–611. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Macdonald A, Campbell DG, Toth R,
McLauchlan H, Hastie CJ and Arthur JS: Pim kinases phosphorylate
multiple sites on Bad and promote 14-3-3 binding and dissociation
from Bcl-XL. BMC Cell Biol. 7:12006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saris CJ, Domen J and Berns A: The pim-1
oncogene encodes two related protein-serine/threonine kinases by
alternative initiation at AUG and CUG. EMBO J. 10:655–664.
1991.PubMed/NCBI
|
|
7
|
Morishita D, Katayama R, Sekimizu K,
Tsuruo T and Fujita N: Pim kinases promote cell cycle progression
by phosphorylating and down-regulating p27Kip1 at the
transcriptional and posttranscriptional levels. Cancer Res.
68:5076–5085. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reeves R, Spies GA, Kiefer M, Barr PJ and
Power M: Primary structure of the putative human oncogene, pim-1.
Gene. 90:303–307. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qian KC, Wang L, Hickey ER, et al:
Structural basis of constitutive activity and a unique nucleotide
binding mode of human Pim-1 kinase. J Biol Chem. 280:6130–6137.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nawijn MC, Alendar A and Berns A: For
better or for worse: the role of Pim oncogenes in tumorigenesis.
Nat Rev Cancer. 11:23–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bullock AN, Debreczeni J, Amos AL, Knapp S
and Turk BE: Structure and substrate specificity of the Pim-1
kinase. J Biol Chem. 280:41675–41682. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mukaida N, Wang YY and Li YY: Roles of
Pim-3, a novel survival kinase, in tumorigenesis. Cancer Sci.
102:1437–1442. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mochizuki T, Kitanaka C, Noguchi K,
Muramatsu T, Asai A and Kuchino Y: Physical and functional
interactions between Pim-1 kinase and Cdc25A phosphatase.
Implications for the Pim-1-mediated activation of the c-Myc
signaling pathway. J Biol Chem. 274:18659–18666. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Z, Bhattacharya N, Mixter PF, Wei W,
Sedivy J and Magnuson NS: Phosphorylation of the cell cycle
inhibitor p21Cip1/WAF1 by Pim-1 kinase. Biochim Biophys Acta.
1593:45–55. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bachmann M, Hennemann H, Xing PX, Hoffmann
I and Möröy T: The oncogenic serine/threonine kinase Pim-1
phosphorylates and inhibits the activity of Cdc25C-associated
kinase 1 (C-TAK1): a novel role for Pim-1 at the G2/M cell cycle
checkpoint. J Biol Chem. 279:48319–48328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bachmann M, Kosan C, Xing PX, Montenarh M,
Hoffmann I and Möröy T: The oncogenic serine/threonine kinase Pim-1
directly phosphorylates and activates the G2/M specific phosphatase
Cdc25C. Int J Biochem Cell Biol. 38:430–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nosaka T and Kitamura T: Pim-1 expression
is sufficient to induce cytokine independence in murine
hematopoietic cells, but is dispensable for BCR-ABL-mediated
transformation. Exp Hematol. 30:697–702. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brault L, Gasser C, Bracher F, Huber K,
Knapp S and Schwaller J: PIM serine/threonine kinases in the
pathogenesis and therapy of hematologic malignancies and solid
cancers. Haematologica. 95:1004–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
20
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Calin GA and Croce CM: MicroRNA-cancer
connection: the beginning of a new tale. Cancer Res. 66:7390–7394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thomas M, Lange-Grünweller K, Weirauch U,
et al: The proto-oncogene Pim-1 is a target of miR-33a. Oncogene.
31:918–928. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grimson A, Farh KK, Johnston WK,
Garrett-Engele P, Lim LP and Bartel DP: MicroRNA targeting
specificity in mammals: determinants beyond seed pairing. Mol Cell.
27:91–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cimmino A, Calin GA, Fabbri M, et al:
miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Calin GA, Dumitru CD, Shimizu M, et al:
Frequent deletions and down-regulation of micro- RNA genes miR15
and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad
Sci USA. 99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bonci D, Coppola V, Musumeci M, et al: The
miR-15a-miR-16-1 cluster controls prostate cancer by targeting
multiple oncogenic activities. Nat Med. 14:1271–1277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Birg F, Courcoul M, Rosnet O, et al:
Expression of the FMS/KIT-like gene FLT3 in human acute leukemias
of the myeloid and lymphoid lineages. Blood. 80:2584–2593.
1992.PubMed/NCBI
|
|
29
|
Nakao M, Yokota S, Iwai T, et al: Internal
tandem duplication of the flt3 gene found in acute myeloid
leukemia. Leukemia. 10:1911–1918. 1996.PubMed/NCBI
|
|
30
|
Yokota S, Kiyoi H, Nakao M, et al:
Internal tandem duplication of the FLT3 gene is preferentially seen
in acute myeloid leukemia and myelodysplastic syndrome among
various hematological malignancies. A study on a large series of
patients and cell lines. Leukemia. 11:1605–1609. 1997. View Article : Google Scholar
|
|
31
|
Thiede C, Steudel C, Mohr B, et al:
Analysis of FLT3-activating mutations in 979 patients with acute
myelogenous leukemia: association with FAB subtypes and
identification of subgroups with poor prognosis. Blood.
99:4326–4335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim KT, Baird K, Ahn JY, et al: Pim-1 is
up-regulated by constitutively activated FLT3 and plays a role in
FLT3-mediated cell survival. Blood. 105:1759–1767. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim KT, Baird K, Davis S, et al:
Constitutive Fms-like tyrosine kinase 3 activation results in
specific changes in gene expression in myeloid leukaemic cells. Br
J Haematol. 138:603–615. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim KT, Carroll AP, Mashkani B, Cairns MJ,
Small D and Scott RJ: MicroRNA-16 is down-regulated in mutated FLT3
expressing murine myeloid FDC-P1 cells and interacts with Pim-1.
PLoS One. 7:e445462012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mizuki M, Fenski R, Halfter H, et al: Flt3
mutations from patients with acute myeloid leukemia induce
transformation of 32D cells mediated by the Ras and STAT5 pathways.
Blood. 96:3907–3914. 2000.
|
|
36
|
Tse KF, Mukherjee G and Small D:
Constitutive activation of FLT3 stimulates multiple intracellular
signal transducers and results in transformation. Leukemia.
14:1766–1776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hayakawa F, Towatari M, Kiyoi H, et al:
Tandem-duplicated Flt3 constitutively activates STAT5 and MAP
kinase and introduces autonomous cell growth in IL-3-dependent cell
lines. Oncogene. 19:624–631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nosaka T, Kawashima T, Misawa K, Ikuta K,
Mui AL and Kitamura T: STAT5 as a molecular regulator of
proliferation, differentiation and apoptosis in hematopoietic
cells. EMBO J. 18:4754–4765. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rayner KJ, Suárez Y, Dávalos A, et al:
MiR-33 contributes to the regulation of cholesterol homeostasis.
Science. 328:1570–1573. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tian Z, Zhao JJ, Tai YT, et al:
Investigational agent MLN9708/2238 targets tumor-suppressor miR33b
in MM cells. Blood. 120:3958–3967. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ibrahim AF, Weirauch U, Thomas M,
Grünweller A, Hartmann RK and Aigner A: MicroRNA replacement
therapy for miR-145 and miR-33a is efficacious in a model of colon
carcinoma. Cancer Res. 71:5214–5224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen JF, Mandel EM, Thomson JM, et al: The
role of microRNA-1 and microRNA-133 in skeletal muscle
proliferation and differentiation. Nat Genet. 38:228–233. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao Y, Ransom JF, Li A, et al:
Dysregulation of cardiogenesis, cardiac conduction, and cell cycle
in mice lacking miRNA-1-2. Cell. 129:303–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Katakami N, Kaneto H, Hao H, et al: Role
of pim-1 in smooth muscle cell proliferation. J Biol Chem.
279:54742–54749. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen J, Yin H, Jiang Y, et al: Induction
of microRNA-1 by myocardin in smooth muscle cells inhibits cell
proliferation. Arterioscler Thromb Vasc Biol. 31:368–375. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Katare RG, Caporali A, Oikawa A, Meloni M,
Emanueli C and Madeddu P: Vitamin B1 analog benfotiamine prevents
diabetes-induced diastolic dysfunction and heart failure through
Akt/Pim-1-mediated survival pathway. Circ Heart Fail. 3:294–305.
2010. View Article : Google Scholar
|
|
47
|
Katare R, Caporali A, Zentilin L, et al:
Intravenous gene therapy with PIM-1 via a cardiotropic viral vector
halts the progression of diabetic cardiomyopathy through promotion
of prosurvival signaling. Circ Res. 108:1238–1251. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mishima T, Mizuguchi Y, Kawahigashi Y and
Takizawa T and Takizawa T: RT-PCR-based analysis of microRNA (miR-1
and -124) expression in mouse CNS. Brain Res. 1131:37–43. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sevignani C, Calin GA, Nnadi SC, et al:
MicroRNA genes are frequently located near mouse cancer
susceptibility loci. Proc Natl Acad Sci USA. 104:8017–8022. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nasser MW, Datta J, Nuovo G, et al:
Down-regulation of micro-RNA-1 (miR-1) in lung cancer. Suppression
of tumorigenic property of lung cancer cells and their
sensitization to doxorubicin-induced apoptosis by miR-1. J Biol
Chem. 283:33394–33405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jin Y, Tong DY, Chen JN, et al:
Overexpression of osteopontin, αvβ3 and Pim-1 associated with
prognostically important clinicopathologic variables in non-small
cell lung cancer. PloS One. 7:e485752012.
|
|
52
|
Nieborowska-Skorska M, Hoser G, Kossev P,
Wasik MA and Skorski T: Complementary functions of the
antiapoptotic protein A1 and serine/threonine kinase pim-1 in the
BCR/ABL-mediated leukemogenesis. Blood. 99:4531–4539. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Eiring AM, Harb JG, Neviani P, et al:
miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation
of mRNA translation in leukemic blasts. Cell. 140:652–665. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kulshreshtha R, Ferracin M, Wojcik SE, et
al: A microRNA signature of hypoxia. Mol Cell Biol. 27:1859–1867.
2007. View Article : Google Scholar
|
|
55
|
Camps C, Buffa FM, Colella S, et al:
hsa-miR-210 Is induced by hypoxia and is an independent prognostic
factor in breast cancer. Clin Cancer Res. 14:1340–1348. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Huang X, Ding L, Bennewith KL, et al:
Hypoxia-inducible mir-210 regulates normoxic gene expression
involved in tumor initiation. Mol Cell. 35:856–867. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kelsey JL: Breast cancer epidemiology:
summary and future directions. Epidemiol Rev. 15:256–263.
1993.PubMed/NCBI
|
|
58
|
Horimoto Y, Hartman J, Millour J, et al:
ERβ1 represses FOXM1 expression through targeting ERα to control
cell proliferation in breast cancer. Am J Pathol. 179:1148–1156.
2011.
|
|
59
|
Malinen M, Jääskeläinen T, Pelkonen M, et
al: Proto-oncogene PIM-1 is a novel estrogen receptor target
associating with high grade breast tumors. Mol Cell Endocrinol.
365:270–276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dumas de la Roque E, Savineau JP and
Bonnet S: Dehydroepiandrosterone: A new treatment for vascular
remodeling diseases including pulmonary arterial hypertension.
Pharmacol Ther. 126:186–199. 2010.PubMed/NCBI
|
|
61
|
Paulin R, Meloche J, Jacob MH, Bisserier
M, Courboulin A and Bonnet S: Dehydroepiandrosterone inhibits the
Src/STAT3 constitutive activation in pulmonary arterial
hypertension. Am J Physiol Heart Circ Physiol. 301:H1798–H1809.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Dhanasekaran SM, Barrette TR, Ghosh D, et
al: Delineation of prognostic biomarkers in prostate cancer.
Nature. 412:822–826. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Guzey M, Kitada S and Reed JC: Apoptosis
induction by 1alpha,25-dihydroxyvitamin D3 in prostate cancer. Mol
Cancer Ther. 1:667–677. 2002.PubMed/NCBI
|
|
64
|
Okamoto R, Delansorne R, Wakimoto N, et
al: Inecalcitol, an analog of 1α,25(OH)(2) D(3), induces growth
arrest of androgen-dependent prostate cancer cells. Int J Cancer.
130:2464–2473. 2012.
|
|
65
|
Ha S, Iqbal NJ, Mita P, et al:
Phosphorylation of the androgen receptor by PIM1 in hormone
refractory prostate cancer. Oncogene. 32:3992–4000. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Maier CJ, Maier RH, Rid R, et al: PIM-1
kinase interacts with the DNA binding domain of the vitamin D
receptor: a further kinase implicated in 1,25-(OH)2D3 signaling.
BMC Mol Biol. 13:182012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shand RL and Gelmann EP: Molecular biology
of prostate-cancer pathogenesis. Curr Opin Urol. 16:123–131. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Roach M III: Current trends for the use of
androgen deprivation therapy in conjunction with radiotherapy for
patients with unfavorable intermediate-risk, high-risk, localized,
and locally advanced prostate cancer. Cancer. Mar 3–2014.(Epub
ahead of print).
|
|
69
|
Carson JP, Kulik G and Weber MJ:
Antiapoptotic signaling in LNCaP prostate cancer cells: a survival
signaling pathway independent of phosphatidylinositol 3′-kinase and
Akt/protein kinase B. Cancer Res. 59:1449–1453. 1999.
|
|
70
|
Sun A, Tang J, Hong Y, et al: Androgen
receptor-dependent regulation of Bcl-xL expression: Implication in
prostate cancer progression. Prostate. 68:453–461. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kumar JK, Ping RY, Teong HF, Goh S and
Clément MV: Activation of a non-genomic Pim-1/Bad-Pser75 module is
required for an efficient pro-survival effect of Bcl-xL induced by
androgen in LNCaP cells. Int J Biochem Cell Biol. 43:594–603. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Min X, Tang J, Wang Y, et al: PI3K-like
kinases restrain Pim gene expression in endothelial cells. J
Huazhong Univ Sci Technolog Med Sci. 32:17–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Muraski JA, Rota M, Misao Y, et al: Pim-1
regulates cardiomyocyte survival downstream of Akt. Nat Med.
13:1467–1475. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zippo A, De Robertis A, Bardelli M,
Galvagni F and Oliviero S: Identification of Flk-1 target genes in
vasculogenesis: Pim-1 is required for endothelial and mural cell
differentiation in vitro. Blood. 103:4536–4544. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Stout BA, Bates ME, Liu LY, Farrington NN
and Bertics PJ: IL-5 and granulocyte-macrophage colony-stimulating
factor activate STAT3 and STAT5 and promote Pim-1 and cyclin D3
protein expression in human eosinophils. J Immunol. 173:6409–6417.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Willert M, Augstein A, Poitz DM,
Schmeisser A, Strasser RH and Braun-Dullaeus RC: Transcriptional
regulation of Pim-1 kinase in vascular smooth muscle cells and its
role for proliferation. Basic Res Cardiol. 105:267–277. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Block KM, Hanke NT, Maine EA and Baker AF:
IL-6 stimulates STAT3 and Pim-1 kinase in pancreatic cancer cell
lines. Pancreas. 41:773–781. 2012.PubMed/NCBI
|
|
78
|
Jacobs MD, Black J, Futer O, et al: Pim-1
ligand-bound structures reveal the mechanism of serine/threonine
kinase inhibition by LY294002. J Biol Chem. 280:13728–13734. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gavara L, Suchaud V, Nauton L, Théry V,
Anizon F and Moreau P: Identification of pyrrolo[2,3-g]indazoles as
new Pim kinase inhibitors. Bioorg Med Chem Lett. 23:2298–2301.
2013.
|
|
80
|
Blanco-Aparicio C and Carnero A: Pim
kinases in cancer: diagnostic, prognostic and treatment
opportunities. Biochem Pharmacol. 85:629–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mumenthaler SM, Ng PY, Hodge A, et al:
Pharmacologic inhibition of Pim kinases alters prostate cancer cell
growth and resensitizes chemoresistant cells to taxanes. Mol Cancer
Ther. 8:2882–2893. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chen LS, Redkar S, Taverna P, Cortes JE
and Gandhi V: Mechanisms of cytotoxicity to Pim kinase inhibitor,
SGI-1776, in acute myeloid leukemia. Blood. 118:693–702. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yang Q, Chen LS, Neelapu SS, Miranda RN,
Medeiros LJ and Gandhi V: Transcription and translation are primary
targets of Pim kinase inhibitor SGI-1776 in mantle cell lymphoma.
Blood. 120:3491–3500. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Batra V, Maris JM, Kang MH, et al: Initial
testing (stage 1) of SGI-1776, a PIM1 kinase inhibitor, by the
pediatric preclinical testing program. Pediatr Blood Cancer.
59:749–752. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen LS, Redkar S, Bearss D, Wierda WG and
Gandhi V: Pim kinase inhibitor, SGI-1776, induces apoptosis in
chronic lymphocytic leukemia cells. Blood. 114:4150–4157. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Chang M, Kanwar N, Feng E, et al: PIM
kinase inhibitors downregulate STAT3(Tyr705) phosphorylation. Mol
Cancer Ther. 9:2478–2487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Siu A, Virtanen C and Jongstra J: PIM
kinase isoform specific regulation of MIG6 expression and EGFR
signaling in prostate cancer cells. Oncotarget. 2:1134–1144.
2011.PubMed/NCBI
|
|
88
|
Xie Y, Burcu M, Linn DE, Qiu Y and Baer
MR: Pim-1 kinase protects P-glycoprotein from degradation and
enables its glycosylation and cell surface expression. Mol
Pharmacol. 78:310–318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Natarajan K, Bhullar J, Shukla S, et al:
The Pim kinase inhibitor SGI-1776 decreases cell surface expression
of P-glycoprotein (ABCB1) and breast cancer resistance protein
(ABCG2) and drug transport by Pim-1-dependent and -independent
mechanisms. Biochem Pharmacol. 85:514–524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kelly KR, Espitia CM, Taverna P, et al:
Targeting PIM kinase activity significantly augments the efficacy
of cytarabine. Br J Haematol. 156:129–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Pogacic V, Bullock AN, Fedorov O, et al:
Structural analysis identifies imidazo[1,2-b]pyridazines as PIM
kinase inhibitors with in vitro antileukemic activity. Cancer Res.
67:6916–6924. 2007.PubMed/NCBI
|
|
92
|
Holder S, Zemskova M, Zhang C, et al:
Characterization of a potent and selective small-molecule inhibitor
of the PIM1 kinase. Mol Cancer Ther. 6:163–172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jie W, He QY, Luo BT, et al: Inhibition of
Pim-1 attenuates the proliferation and migration in nasopharyngeal
carcinoma cells. Asian Pac J Trop Med. 5:645–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Akué-Gédu R, Rossignol E, Azzaro S, et al:
Synthesis, kinase inhibitory potencies, and in vitro
antiproliferative evaluation of new Pim kinase inhibitors. J Med
Chem. 52:6369–6381. 2009.PubMed/NCBI
|
|
95
|
Santio NM, Vahakoski RL, Rainio EM, et al:
Pim-selective inhibitor DHPCC-9 reveals Pim kinases as potent
stimulators of cancer cell migration and invasion. Mol Cancer.
9:2792010. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Mori M, Tintori C, Christopher RS, et al:
A combination strategy to inhibit Pim-1: synergism between
noncompetitive and ATP-competitive inhibitors. ChemMedChem.
8:484–496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Grundler R, Brault L, Gasser C, et al:
Dissection of PIM serine/threonine kinases in FLT3-ITD-induced
leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated
homing and migration. J Exp Med. 206:1957–1970. 2009. View Article : Google Scholar : PubMed/NCBI
|