Introduction
Esophageal carcinoma (EC) is one of the most lethal
types of digestive tract malignancy according to 2011 cancer
statistics, and there is clear geographic variation in its
incidence throughout the world (1). Esophageal adenocarcinoma (EAC) and
esophageal squamous cell carcinoma (ESCC) are the most common
histopathological types of EC. EAC almost uniquely
histopathologically features in western countries, but ESCC is the
most common type in Asian countries such as China and Japan
(1). Similar to the
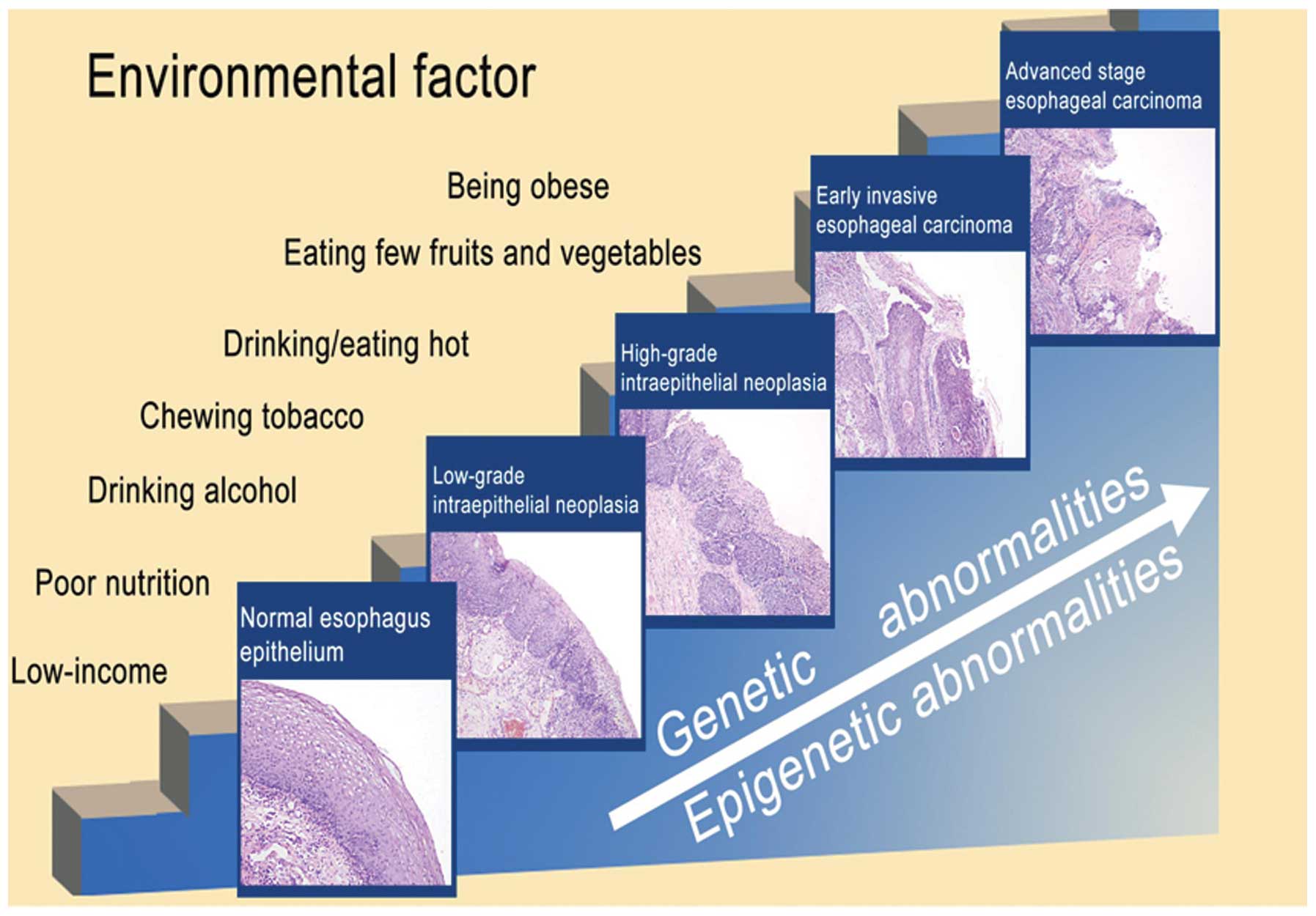
adenoma-carcinoma sequence of EAC (2), ESCC develops through progression from
normal esophageal epithelium (NEE) to low-grade intraepithelial
neoplasia (LGIN), high-grade intraepithelial neoplasia (HGIN),
early stage esophageal carcinoma (EEC) and then advanced stage
esophageal carcinoma (AEC) with an accumulation of genetic and
epigenetic abnormalities (Fig. 1).
Studies on esophageal precancerous lesions, including Barrett’s
esophagus and EAC have produced valuable results (3) and a number of biomarkers have entered
clinical trials in the USA (2).
Other studies have also investigated precancerous lesions (3–5);
however, further research is required. In China, a large-sample
study demonstrated that the canceration rate in NEE was 1.4%, while
the canceration rates of LGIN and HGIN were 5.8 and 38.9%,
respectively (6). These results
demonstrate that it is essential to identify novel molecular
markers in precancerous patients that are at high risk of a
malignant transformation in order to prevent the development of
ESCC.
Historically, mild, moderate and severe dysplasia
were the terms used to describe premalignant squamous epithelium
cellular changes. Although it remains in use, this nomenclature has
generally been replaced by the term of esophageal intraepithelial
neoplasia (EIN), which is used to describe histological changes
that are detected with biopsy. EIN, also known as esophageal
precancerous conditions (EPC), is the potentially premalignant
transformation and abnormal growth of squamous cells on the surface
of the esophagus. Previous studies have confirmed that EIN was a
definite risk factor for ESCC (4,5,7).
Several novel genes associated with EIN and their molecular
mechanisms of suppression, or even activation, have been detected.
Kamangar et al (8)
demonstrated that serum pepsinogen I (PGI) had no statistically
significant association with EIN, whether analyzed as a
dichotomous, ordinal (quartiles), or continuous variable, but a
lower serum PG I/II ratio was linearly associated with higher risk
of EIN. Chen et al (9) also
confirmed that serum matrix metalloprotease-9 had a statistically
different distribution between no dysplasia (normal and
esophagitis) and dysplasia/early cancer subjects, yet this
biomarker exhibited poor performance in a subsequent screening test
and displayed low sensitivity and specificity. In cancerous tissues
and precancerous lesions, Kobayashi et al (5) showed that p53 point mutation was
involved in esophageal carcinogenesis. Another study reported that
cyclin D1 overexpression starts early in dysplasia and could be a
useful marker for its malignant potentiality, while reduction of
p16INK4 and p27KIP1 expression occurs during the transformation
from dysplasia to cancer (10).
These findings suggested that numerous genes are abnormally
expressed in EIN, which should be treated as a precancerous lesion.
A previous study demonstrated that Ki-67 and ProExC can be used as
an adjunct tool for diagnosing difficult cases of EIN (11). Another previous study showed that
reduction of NOTCH1 expression directs the basal cells to cease
terminal differentiation and to form an immature epithelium,
thereby exhibiting a major role in the histopathogenesis of
squamous epithelium neoplasia (12), but its expression and function in
esophageal epithelium has not been investigated to the best of our
knowledge.
The majority of the transcriptional output of the
mammalian genome has been confirmed to be non-protein-coding
(13), and these abundant parts of
the transcriptome, which were previously regarded as
‘transcriptional noise’, have been identified to have important
regulatory potential in transcription and post-transcription
(14,15). Non-coding RNA (ncRNA) is a type of
RNA that does not code for protein but has enzymatic, structural or
regulatory function (16). ncRNAs
can be classed as either small or long ncRNA, based on their
transcript length (17). The most
studied class of short ncRNA is microRNA (miRNA), which is involved
in the specific regulation of its target messenger RNAs (mRNAs)
through the inhibition of post-transcriptional cleavage or
translation (18). Studies have
demonstrated the differential expression patterns of miRNAs in
numerous types of cancer. MiR-92a (19), miR-103/107 (20), miR-21, miR143, miR145, miR-205
(21) and miR-296 (22), among others, have been confirmed to
be involved in the development of ESCC. lncRNA is a novel class of
ncRNAs that are >200 nucleotides in length. Despite no known
protein-coding potential, these RNAs demonstrate a wide range of
structural and functional roles in various processes, including
imprinting control (23), cell
differentiation (24,25), immune responses (26,27),
tumorigenesis (28–31), memory (32) and determination of pluripotency of
embryonic stem cells (33).
Large-scale analyses of full-length cDNA sequences have detected
numerous lncRNAs in humans (34,35)
and mice (36), but only a small
number of these nucleic acids have been well characterized
functionally. A number of studies have produced data implying that
the effects of lncRNAs and their mechanisms of gene expression and
regulation may be much broader and more complex than those of
miRNAs (37–39). The development of
molecular-profiling techniques, such as cDNA microarrays and
transcriptome sequencing, may facilitate the provision of gene
panels that identify EIN- and ESCC-specific molecular patterns. In
a previous study, the upregulation of the lncRNA, HOTAIR, which was
originally discovered in breast cancer tissues, was demonstrated to
promote cancer metastasis and predict poor prognosis in ESCC
(40). However, there are few
studies concerning the expression profiles and functions of lncRNAs
in esophageal diseases (41,42).
In the present study, ncRNA and mRNA expression
profiles in EIN and ESCC samples were compared with those in NEE
tissues using microarrays. To the best of out knowledge, this is
the first study to determine the expression patterns of genome-wide
miRNAs, mRNAs and lncRNAs in canceration processes of ESCC by
microarray.
Materials and methods
Sample collection
The present study was approved by the Institutional
Review Boards of the Cancer Center, Affiliated Nanjing Hospital of
Nanjing Medical University (Nanjing, China), and all subjects
provided written informed consent. All samples were obtained from
the Nanjing Hospital (Nanjing, China), between January and
September 2011. Biopsy specimens (NEE, LGIN, and HGIN samples) were
obtained via esophageal endoscopy. Two biopsy samples were
collected at once; the first biopsy tissue sample was routinely
sent for pathological diagnosis, and then the pathological results
determined whether the second sample was suitable for study.
Unstained or lightly stained areas following Lugol’s iodine
staining were considered to be precancerous lesions. EEC and AEC
tissues were procured following surgical resection. Biopsies and
surgical specimens were then placed in RNase-free freezer tubes
(1.2 ml; Corning, Inc., Corning, NY, USA) and snap-frozen in liquid
nitrogen.
Histopathological analysis
Terminology
NEE denotes normal esophageal squamous epithelium,
with no esophagitis, basal cell hyperplasia, or other abnormal
conditions. LGIN denotes a low- or moderate-grade lesion. It refers
to mild/moderate atypical cellular changes which are confined to
the lower third or the basal two-thirds of the epithelium. HGIN
denotes a high-grade lesion. It refers to severe atypical cellular
changes spanning more than two-thirds of the epithelial thickness,
including full-thickness lesions. Carcinoma in situ is a
pathological type of HGIN, but much more severe. EEC is the primary
tumor, which is confined to the adventitia of the esophagus, with
no metastasis to the lymph nodes or distant organs. AEC refers to a
primary tumor that has spread beyond the adventitia of the
esophagus, with metastasis present in the lymph nodes or distant
organs.
Biopsy samples
Twenty-three patients underwent esophageal
endoscopy, and their biopsy samples were diagnosed based on the
World Health Organization International Classification of Diseases
for Oncology (2010). Five cases of NEE, four cases of LGIN and two
cases of HGIN were diagnosed. A number of the subjects were
diagnosed with esophagitis (n=8), or basal cell hyperplasia (n=4)
without any other diagnoses of greater severity, so these subjects
were excluded from further analysis.
Surgical samples
Among the twenty patients from whom samples were
collected, three patients were discovered to have adenocarcinoma or
were undergoing preoperative treatment, and eight samples were
classed as well and poorly differentiated, so were excluded from
further analysis. After exclusions, nine patients who had undergone
no preoperative treatment remained, including seven cases of EEC
(moderate differentiation), and two cases of AEC (with moderate
differentiation).
All samples were selected based on their
pathological diagnosis and then reviewed by another pathologist to
ensure correct diagnoses. The clinicopathological features of the
20 patients included in the study were reported according to the
pathological tumor-node-metastasis classification of the
International Union Against Cancer (Seventh edition). The 5
patients who displayed no evidence of disease were selected as
controls and were matched to the patients by age, gender and
ethnicity (Table I).
 | Table ICharacteristics of patient
tissues. |
Table I
Characteristics of patient
tissues.
| Sample number | Age | Gender | Pathological
diagnosis |
|---|
| 1 | 56 | M | NEE |
| 2 | 56 | M | NEE |
| 3 | 60 | M | NEE |
| 4 | 38 | F | NEE |
| 5 | 59 | F | NEE |
| 6 | 57 | F | LGIN |
| 7 | 69 | M | LGIN |
| 8 | 58 | F | LGIN |
| 9 | 59 | F | LGIN |
| 10 | 65 | M | HGIN |
| 11 | 52 | M | carcinoma in
situ |
| 12 | 76 | M | T3N0/20M0 |
| 13 | 62 | M | T2N0/30M0 |
| 14 | 73 | M | T3N0/11M0 |
| 15 | 69 | M | T3N0/31M0 |
| 16 | 55 | F | T3N0/12M0 |
| 17 | 59 | F | T3N0/17M0 |
| 18 | 59 | M | T3N0/18M0 |
| 19 | 60 | M | T3N5/15M0 |
| 20 | 72 | F | T3N1/33M0 |
RNA extraction and quality
monitoring
Biopsy samples were subjected to RNA extraction
using the RNeasy Micro kit (Qiagen, Valencia, CA, USA) according to
the manufacturer’s instructions. TRIzol reagent (Invitrogen,
Carlsbad, CA, USA) was used according to the manufacturer’s
instructions with minor modification in order to extract total RNA
from the surgical samples. The aqueous phase was subjected to 3
steps of acid phenol/chloroform purification to eliminate protein
residues prior to isopropyl alcohol precipitation. The resulting
RNA pellet was then dissolved in 5 or 40 μl
diethylpyrocarbonate-treated water. The RNA integrity was evaluated
with a NanoDrop ND-2000 spectrophotometer (Thermo Fisher
Scientific, Wilmington, DE, USA) and standard denaturing agarose
gel electrophoresis.
For the microarray, equal volumes of total RNA from
each of the patients were pooled separately in accordance with the
RNA concentration of each sample to form disease and control sample
pools.
miRNA and lncRNA-mRNA microarrays
miRNAs
The 5 groups of samples were separately labeled
using the miRCURY LNA miRNA Hy3/Hy5 Power Labeling kit (208030-A;
Exiqon, Woburn, MA, USA) and hybridized on the miRCURY LNA miRNA
array (version 14.0; 5th Gen Human; Exiqon). After the washing
steps, the slides were scanned using the Axon GenePix 4000B
Microarray scanner (Molecular Devices, Sunnyvale, CA, USA). Scanned
images were then imported into GenePix Pro 6.0 software (Axon;
Molecular Devices) for grid alignment and data extraction.
Replicated miRNAs were averaged and miRNAs with intensities >50
in all samples were selected for calculating the normalization
factor. Expressed data were normalized using the median. Following
normalization, differentially expressed miRNAs were identified
through fold change filtering.
lncRNAs and mRNAs
A Human lncRNA array version 1.0 (12×135k;
Arraystar, Shanghai, China), containing probes for 18,534 lncRNAs
and 18,874 coding transcripts (collected from databases such as
NCBI RefSeq, UCSC Known Genes, NRED, RNAdb and Ensembl), was used
for detection. Total RNA (~5 μg) from each sample was used for
labeling and array hybridization with the following steps: i)
Reverse transcription with a SuperScript Double-Stranded cDNA
Synthesis kit (Invitrogen); ii) double-stranded cDNA labeling with
a NimbleGen One-Color DNA Labeling kit (Roche, Mannheim, Germany);
iii) array hybridization using the NimbleGen Hybridization System
(Roche), followed by washing with the Nimblegen Wash Buffer kit
(cat. no. 05584507001; Roche); and iv) array scanning using the
Axon GenePix 4000B Microarray scanner (Molecular Devices). Scanned
images (TIFF format) were then imported into NimbleScan software
(version 2.5; Roche) for grid alignment and expression data
analysis. Additionally, hierarchical clustering was performed to
present distinguishable mRNA expression profiling among
samples.
Bioinformatic analysis
All data were divided into three groups (lncRNA,
miRNA and mRNA), and each group contained five stages. Stage 1
(NEE) is the normal control condition, while stages 2 (LGIN), 3
(HGIN), 4 (EEC), and 5 (AEC) were the disease stages. The period
between stages 3 and 4 is the most important transition period, so
the dysregulation and potential functions of miRNAs, mRNAs and
lncRNAs in these two stages were carefully analyzed.
Data preprocessing
Expression values of miRNAs, mRNAs and lncRNAs that
were expressed in the five stages were selected (Table 1, Supplementary Material, http://210.46.85.200/Supplemental-Material/download.jsp),
and differential expression values of the three categories of RNA
were identified by screening for genes that were differentially
expressed in the different disease stages compared with the normal
group (Table 2, Supplementary Material).
Two-fold changes was selected as the minimum difference threshold.
Two collections of differentially expressed genes were extracted:
One consisted of those that were differentially expressed between
the normal group and all of the four disease phases (intersection),
and the other included all of the genes that were differentially
expressed between the normal group and any of the disease stages
(union) (Table 1Supplementary Table 2.–3,
Supplementary Material).
Data analysis
The following aspects were analyzed: (i) The extent
of overlapping expression between the two collections and the
clustering of the union and intersection differentially expressed
RNAs were evaluated based on their fold change. (ii) The
differential expression patterns of lncRNA, miRNA and mRNA at
stages 3 and 4 vs. the normal group. The following sets of data
were used for this analysis: The lncRNAs which were downregulated
in stage 3 and upregulated in stage 4 vs. the normal group; the
miRNAs which were significantly upregulated in stages 3 and 4 vs.
the normal group; and the mRNAs which were downregulated in stages
3 and 4 vs. the normal group. The latter two sets of data were used
to analyze the association between these reduced mRNAs and
upregulated miRNAs. (iii) The similarities of the potential
functions of miRNAs, mRNAs and lncRNAs in the initiation and
progression of ESCC. a) Similarities between the miRNAs and mRNAs.
The clustering analysis of the union and intersection sets of
differentially expressed miRNAs, and the target mRNAs of these
miRNAs were acquired. mRNAs that had significantly downregulated
expression levels in stages 3 and 4 vs. the normal group were
selected for further analysis. Then, the mRNAs that were both
targets of the miRNAs and downregulated in the microarray during
this period were selected. All of these selected mRNAs were
subjected to GO analysis, and then all related miRNAs underwent
functional enrichment by DAVID (http://david.abcc.ncifcrf.gov). b) Similarities
between the mRNAs and lncRNAs. The lncRNAs with significantly
upregulated expression levels in both stages 3 and 4 vs. the normal
group were selected for further analysis. The two neighboring mRNAs
of a lncRNA were used to define the function of the lncRNA. All
related mRNAs were subjected to GO analysis, and then all related
lncRNAs underwent functional enrichment by DAVID. c) Similarities
between the miRNAs, mRNAs and lncRNAs. Functional similarities
between the miRNAs and mRNAs, and the lncRNAs and mRNAs were
compared in order to determine common features of the three types
of RNA in the canceration processes of ESCC.
Results
Identification of lncRNAs, miRNAs and
mRNAs dysregulated during the progression of esophageal
carcinoma
The detection rates of the expression of miRNAs,
mRNAs and lncRNAs were 3.74 (59/1,700), 71.61 (13,517/18,874) and
77.14% (14,298/18,534), respectively. The results indicated that
4,404 lncRNAs, 36 miRNAs and 12,872 mRNAs were co-expressed in all
five stages. Compared with the NEE (stage 1), the numbers of
differentially expressed RNAs in the 4 disease stages (stages 2–5)
were as follows: lncRNAs, 2,390, 1,567, 2,961 and 2,016; miRNAs,
15, 18, 14 and 17; and mRNAs, 6,370, 4,768, 7,229 and 4,959,
respectively. The co-expressed and differentially expressed genes
were compared, and 435 lncRNAs (Fig.
2A), 7 miRNAs (Fig. 2B), and
1,265 mRNAs (Fig. 2C) were
expressed in all 5 stages and were differentially expressed in
stages 2–5 vs. the normal group (Table 1Supplementary Table 2.Supplementary Table 3.–4,
Supplementary Material).
Similarities of lncRNAs, miRNAs and mRNAs
in dysregulated processes of esophageal carcinoma
ESCC is characterized by its aggressiveness and poor
prognosis, and frequently develops from varying degrees of IN (LGIN
to HGIN), which is a premalignant pathological condition occurring
in normal esophagi. A number of studies have confirmed that HGIN is
the most common precancerous lesion and often advances to ESCC
(3–12). Therefore, the present study focused
on the HGIN to EEC period (stages 3–4), which is the most important
transition period.
Clustering analysis of the intersection (Fig. 3A, bottom) and union (Fig. 3A, top) sets of differentially
expressed lncRNAs in the four stages of disease (compared with
stage 1) were constructed based on the fold change. The results
displayed that expression levels of the majority of lncRNAs were
upregulated (Table 5, Supplementary
Material).
Additionally, clustering analysis of the
intersection (Fig. 3B, bottom) and
union (Fig. 3B, top) sets of
differentially expressed miRNAs in the 4 disease stages were
constructed based on the fold change. In the clustering map of the
union set of differentially expressed miRNAs, only 2 miRNAs
(miR361-3p and miR1470) were significantly upregulated in stages 3
and 4.
Similarities between miRNAs and
mRNAs
miRNAs are endogenous small ncRNAs that are ~22 nt
in length. They negatively regulate gene expression by binding to
the 3′-untranslated regions (UTRs) of mRNA target transcripts,
causing translational repression or mRNA degradation. As miR361-3p
and miR1470 were indicated to be significantly upregulated in
stages 3 and 4, the target mRNAs of these two miRNAs were analyzed,
and the target data sets of the two miRNAs were obtained from
target gene prediction databases, including TargetScan (www.targetscan.org), miRBase (www.mirbase.org) and miRanda (http://www.microrna.org). There were 5,247 target
mRNAs of miR361-3p, and 526 target mRNAs of miR1470 (Table 6, Supplementary Material). Based on the fold
change, clustering analysis of the intersection (Fig. 3C, bottom) and union (Fig. 3C, top) sets of differentially
expressed mRNAs in the four stages of disease was performed. The
expression analysis of mRNAs was somewhat indiscriminate,
exhibiting patterns of upregulation and downregulation in stages 3
and 4. In order to study the association between miRNAs (increased
expression in stages 3 and 4 vs. the normal group) and mRNAs, 2,943
mRNAs that were downregulated in stages 3 and 4 vs. the normal
group were selected (Table 7, Supplementary
Material). mRNAs that were both predicted targets of the miRNAs and
downregulated in stages 3 and 4 were selected and identified as
miRNA-associated mRNAs (intersection). A total of 642 target mRNAs
of miR361-3p, and 71 target mRNAs of miR1470 were identified as
miRNA-associated mRNAs (Table 8, Supplementary
Material). All selected mRNAs in this intersection set underwent GO
analysis, and 139 GO terms were acquired following functional
enrichment analysis by DAVID (Table 9,
Supplementary Material). According to the functional enrichment
analysis of the mRNAs, 26 GO terms were involved in aspects of
cancer, including the cell cycle, cell death, cell communication,
signal transduction and apoptotic process. Results of the current
study further supported the hypothesis that these dysregulated
molecules may be involved in the complicated associations in ESCC
development.
Link between lncRNAs and mRNAs
The possible function of lncRNAs was probed
according to neighboring (upstream and downstream) mRNAs in the
current study. The neighboring mRNAs of lncRNA, which displayed
significantly increased expression in stages 3 and 4 vs. the normal
group were collected. In the present study, 1,887 lncRNAs with
neighboring mRNAs were identified and all the related mRNAs
underwent GO analysis and functional enrichment by DAVID (Table 10, Supplementary Material). The results
indicated that these associated lncRNAs and mRNAs were involved in
apoptosis, the cell cycle, proliferation, invasion and metastasis,
which encompass the majority of the regulatory processes in the
biological behavior of tumor cells.
Functions shared by lncRNAs, miRNAs and
mRNAs
The mRNAs that were both targets of miR361-3p or
miR1470 and downregulated in stages 3 and 4 were obtained.
Subsequently, the set of intersecting lncRNAs-mRNAs and
miRNAs-mRNAs was selected and the similar potential functions among
the miRNAs, mRNAs and lncRNAs in initiation and progression of ESCC
were acquired (Table 11, Supplementary
Material). Based on the analysis, the cross-linked diagrams of
miR361-3p and miR1470 are depicted in Fig. 4; the three types of RNA shared
similarities in the majority of the stages of disease progression.
The function of these cross-linked lncRNAs, miRNAs and mRNAs in
carcinogenesis and development of ESCC was then analyzed. The
cluster analyses of the differentially expressed lncRNAs, miRNAs
and mRNAs (disease stages vs. the normal group) are displayed in
Fig. 5. The GO functional
enrichment of the intersection set of mRNAs was then acquired. For
example, the GO term 0008219 is correlated with cell death, and
nine mRNAs (NM_171982, NM_022470, NM_145725, NM_000332,
NM_001098517, NM_001004426, NM_152240, NM_002598 and NM_004394)
that were downregulated in the disease and may be regulated by
miR-361-3p were enriched in this GO term, while almost eight
lncRNAs that neighbored the mRNAs were also enriched in the same GO
term (Table 12, Supplementary Material). These
results suggested that these lncRNAs, mRNAs and miRNAs may share
similar potential functions in the regulation of cell death,
possibly in apoptosis, cell cycle, invasion and metastasis, through
which they promote the tumorigenesis and development of ESCC.
Therefore, this study revealed the complicated interlinked
functions of lncRNAs, miRNAs and mRNAs, and partially confirmed the
regulation of the molecular network in ESCC (Table
12, Supplementary Material).
Discussion
Malignant transformation from NEE to ESCC is a
multistep process involving an accumulation of genetic and
epigenetic changes. However, the mechanism of the development of
ESSC remains unclear. Cell malignant transformation may be
influenced by genetic background and environmental factors,
including poor nutrition, unhealthy diet, smoking, drinking
alcohol, and obesity. A previous study hinted that molecular
changes may occur prior to histomorphological changes in the
occurrence of cancer (43). The
concept of the functional genome now includes a multitude of newly
discovered classes of ncRNA transcript (16). Although the functional significance
of ncRNAs has long been recognized, the abundance and scale of
ncRNA (particularly lncRNA) expression changes in cancer is just
beginning to become clearer. Thus far, few studies of the
expression profiles and functions of lncRNA exist. It has been
demonstrated that a novel lncRNA, HOTAIR was involved in various
types of tumor. In a previous study, the lncRNA HOTAIR was
upregulated and promoted cancer metastasis and predicted poor
prognosis in ESCC (40). At
present, the function of the majority of lncRNAs in ESCC is not
clear. For this reason, charting the transcriptional landscape of
coding and ncRNAs across normal and disease tissues is a key step
in understanding the functional significance of transcriptome
regulation in ESCC.
In the current study, to the best of our knowledge,
an analysis of the stages of ESCC, human tissue-associated ncRNA
and mRNA expression profiling was presented for the first time.
Five lncRNA-mRNA microarrays and five miRNA microarrays were used
to detect five NEE, four LGIN, two HGIN, seven EEC and two AEC
tissues. However, the single size of each biopsy sample was too
small to extract sufficient RNA to meet the requirements of the
microarray, therefore, this study adopted a mixed-sample method,
and the effective information of each individual could not be
ascertained.
A first generation atlas of the expression profiles
of coding and non-coding RNA has been produced in the present
study, providing novel insight for this rapidly growing area of
research into precancerous and cancerous diseases. Analysis in the
present study indicated that 7 miRNAs, 1,265 mRNAs, and 435 lncRNAs
exhibited differential expression between normal and disease
tissues. The findings suggest that the majority of genes
dysregulated during disease progression of ESCC influence the
progression from the normal state to the IN and cancerous states. A
high percentage of genes were dysregulated during the progression
from HGIN to invasive cancer. The number of differentially
expressed miRNAs was lower than that of mRNAs and lncRNAs in the
stages 2–5, when compared with stage 1. It is also possible that
the differences were due to the total number of miRNAs being less
than those of mRNAs and lncRNAs. Although the total number of
miRNAs in the intersection set was relatively small, it has been
previously demonstrated that miRNAs are important in the regulation
of the expression of their target mRNAs, thus confirming that
miRNAs also have important roles in the tumorigenesis and
development of esophageal diseases (44). In the present study, miRNAs,
lncRNAs and mRNAs were demonstrated to be differentially expressed
in the disease stages compared with the normal group, indicating
that these types of molecules are involved in the tumorigenesis and
development of cancer. It was also confirmed that a large number of
the lncRNAs and mRNAs which were associated in the disease stages
may be important in esophageal carcinogenesis. However, the
distribution pattern of miRNAs did not correlate with that of
protein-coding genes or lncRNAs.
Several studies have now indicated that lncRNAs
regulate gene expression in cis or trans and may also function as
transcriptional enhancers (45).
Wamstad et al (46)
hypothesized that if lncRNAs function in cis to regulate lineage
commitment, then their neighboring genes should have functions
associated with this process. To test this theory, they determined
GO enrichment for the two nearest genes relative to the lncRNAs.
Consistent with their hypothesis, they noted enrichment of genes
involved in development, morphogenesis, and transcriptional
processes. They found that lncRNAs identified in the data were
significantly correlated in expression with their neighboring genes
compared with randomly selected neighboring protein-coding genes.
They tested the possibility that the observed correlations are
attributable to coordinately regulated gene clusters; however, they
observed that lncRNA expression is more highly correlated with the
nearest adjacent gene (P=0.0275) relative to their background
model.
Studies have demonstrated that lncRNAs perform their
functions of gene regulation through interaction with their
adjacent transcripts in cis. X-chromosome inactivation in mammals
relies on XIST, a long non-coding transcript that coats and
silences the X chromosome in cis. Vallot et al (47) reported the discovery of another
lncRNA, XACT, which was expressed from and coated the active X
chromosome specifically in human pluripotent cells, and in the
absence of XIST, XACT was expressed from the two X chromosomes in
humans but not in mice, suggesting a unique role for XACT in the
control of human XCI initiation. Another paternally expressed
lncRNA termed Kcnq1ot1 regulates epigenetic gene silencing in an
imprinted gene cluster in cis over a distance of 400 kb in the
mouse embryo, whereas the silenced region extends over 780 kb in
the placenta (48). Furthermore, a
large ncRNA called ANRIL (for antisense noncoding RNA in the INK4
locus) has been identified within the p15/CDKN2B-p16/CDKN2Ap14/ARF
gene cluster. Genome-wide association studies also identified ANRIL
as a risk locus for gliomas and basal cell carcinomas.
Additionally, a mouse model has confirmed the pivotal role of ANRIL
in regulation of CDKN2A/B expression through a cis-acting mechanism
and its implication in proliferation and senescence (49). These results were consistent with
the findings of Wamstad et al and further confirmed that it
was sensible to probe the function of an unknown lncRNA based on
the style of cis-regulation. Cis regulation is part of the
mechanism of lncRNA gene regulation; however, thus far it has
proven difficult to ascertain the cis or trans regulation manner of
a specific lncRNA. In the current study, the cis analytical method
was used, which partly revealed the function of lncRNAs in the
initiation and progression of ESCC. According to the functional
comparison of the association of lncRNA-mRNA and miRNA-mRNA,
similarities in the potential functions of them in the
carcinogenesis of esophageal squamous cells were identified.
Consistent with this theory, methylation regulation (50) and ‘sponge adsorption’ theory
(51), functional cross-linking
among the three types of molecule has been demonstrated in the
pathological process of cancer. Braconi et al (50) highlighted the inter-association
between two classes of ncRNA, miRNA and lncRNA, and revealed
miRNA-dependent regulation of lncRNA MEG3 expression by evaluating
the involvement of miR-29, which can modulate DNMT 1 and 3. Their
findings showed that overexpression of mir-29a increased the
expression levels of MEG3. These data showed that
methylation-dependent tissue-specific regulation of the lncRNA MEG3
by miR-29a may contribute to HCC growth. Wang et al
(51) also demonstrated that the
lncRNA HULC may act as an endogenous ‘sponge’, which downregulates
miR-372 activity, and inhibition of miR-372 leads to reducing
translational repression of its target gene, PRKACB, which in turn
induces phosphorylation of CREB and HULC expression. Their data
elucidated that fine tuning of the expression of the lncRNA HULC is
part of an auto-regulatory loop, in which the inhibitory effect of
HULC on the expression and activity of miR-372 allows upregulated
expression of HULC in liver cancer. Collectively, these studies
have identified a diverse repertoire of ncRNA functions but may
have only scratched the surface of the functions of lncRNAs in
cancer. Follow-up studies are required to reveal the related
functions among lncRNAs, miRNAs and mRNAs in ESCC. Certainly,
further regulatory mechanisms and related pathways remain
undiscovered. Future studies should aim to pinpoint potential
functions of lncRNAs and discern whether the non-coding genome
contributes to ESCC, and the mechanisms by which it does this.
A total of twenty-six GO terms were observed in the
present study, of which miR-361-3p and miR-1470 shared similarities
in GO functional enrichment involved in cancer, including cell
cycle, cell death, cell communication, signal transduction and
apoptotic process. These miRNAs have been confirmed to have
important roles in numerous types of tumor. miR-361-3p is a highly
conserved X-linked miRNA that was demonstrated to be dysregulated
in the serum of patients with lung cancer, and may be a blood-based
marker for discriminating between malignant and benign lung tumors
(52). Tanaka et al
(53) reported that miR-361-3p was
an oncogenic miRNA in human oral cancer cells. Hughes et al
(54) confirmed that miR-361-3p
expression was significantly increased in clear cell renal cell
carcinoma compared with that in normal renal tissues. miR1470 has
been demonstrated as another tumor-associated miRNA; Xiong et
al (55) demonstrated that
miR-1470 was one of known leukemia-associated miRNAs. A study by
Sultan et al (56) also
supported the theory by demonstrating miR-1470 overexpression in
paclitaxel-resistant ovarian cancer cell lines compared with
parental cell lines. The findings of this study provide novel
insight into the carcinogenesis process of esophageal squamous
cells, particularly regarding precancerous lesions. This study
could also potentially be the basis of new predictive biomarkers in
the future and aid improvement of the understanding of malignant
transformation development.
Due to constraints of sample acquisition and
research conditions, further experiments were not performed to
validate the results of the present study. However, a number of
previous studies have partially confirmed the results of this
analysis. A number of the tumor-associated mRNAs that have been
previously analyzed have been demonstrated to be involved in the
initiation and progression of ESCC. For example, CDC25B was
revealed to be an oncogene, influencing G2-M progression (57). Xue et al (58) analyzed the protein expression
patterns of CDC25B and concluded that it would be valuable for the
development of rational strategies for early detection of lesions
that may lead to advanced ESCC. Furthermore, Li et al
(59) demonstrated the expression
levels of CDC25B in esophageal carcinoma to be significantly higher
than those in dysplasia and normal tissues (48.1, 16 and 0%,
respectively, P<0.01), and that CDC25B expression was correlated
with the degree of differentiation and depth of invasion of tumor
cells. Thus, CDC25B may be important in the early phase of ESCC. In
addition, Jiang et al (60)
demonstrated that the levels of ATM expression were increased in
ESCC and premalignant lesions compared with those in normal tissues
using in situ hybridization, and that increased ATM
expression levels were associated with tumor de-differentiation.
Their findings also suggested that the ATM gene should be further
evaluated as a biomarker for the early detection of esophageal
cancer in patients. DAPK1 is another associated gene that may
participate in metastasis and apoptosis of ESCC cells, and its
protein expression is closely correlated with the
clinicopathological characteristics of ESCC (61). Currently, it is considered that EIN
is a precancerous stage of esophageal cancer, and mounting evidence
supports chronic inflammation as one of the promoters of EIN
formation. BIRC2 was one of the inflammatory carcinogenesis-related
genes in the present study. Daigeler et al (62) displayed that BIRC2 was upregulated
in ESCC, while Fukuda et al (63) have reported that upregulated BIRC2
is associated with apoptosis and the inflammatory response. At
present, EIN is considered to be a precancerous stage of esophageal
cancer. Numerous studies have confirmed that chronic inflammation
was one of the important factors promoting EIN formation. The
aforementioned studies have shown that these dysregulated mRNAs may
have important roles in the pathological process of ESCC. However,
the regulatory mechanism of dysregulated mRNAs remains to be
elucidated. Findings of the current study may provide novel insight
into the processes of esophageal squamous cell carcinogenesis,
particularly concerning precancerous lesions. Further studies are
warranted to determine the functional role of these transcripts
during the initiation and progression of esophageal tumors.
EIN is a potentially precancerous lesion, which is
diagnosed histopathologically. Currently, only histological
alterations are of practical value in routine clinical settings and
histological diagnosis is the gold standard for surveillance of
patients with IN. However, it is not presently possible to predict
which lesions will progress. The combination of clinical parameters
and genetic/epigenetic alterations increases the quality of the
risk assessment of ESCC in IN patients. Identification of specific
ncRNA patterns and their association with mRNAs in canceration
processes of ESCC will help distinguish high-risk from low-risk
patients. These potential biomarkers may be proteins or genes that
can be differentially expressed in cancer, precancer and normal
tissue.
The data of the present study demonstrate the
similarities of lncRNAs, miRNAs and mRNAs in the initiation and
progression of ESCC and provide novel insight for this rapidly
growing area of research into precancer and cancer of the
esophagus.
Supplementary Materials
Acknowledgements
This study was supported by the Graduate Research
and Innovation Projects of Jiangsu (grant no. CXLX_0567), the
Foundation of Nanjing City Committee of Science and Technology
(grant no. 201108027), the Innovation Team of Science and Education
Health Project of Jiangsu in 2011 (grant no. 15) and the National
Natural Science Foundation of China (grant no. H1617/81201881).
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Tischoff I and Tannapfel A: Barrett’s
esophagus: can biomarkers predict progression to malignancy? Expert
Rev Gastroenterol Hepatol. 2:653–663. 2008.
|
|
3
|
Kaneko K, Katagiri A, Konishi K, et al:
Study of p53 gene alteration as a biomarker to evaluate the
malignant risk of Lugol-unstained lesion with non-dysplasia in the
oesophagus. Br J Cancer. 96:492–498. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shimizu Y, Yoshida T, Kato M, et al:
Low-grade dysplasia component in early invasive squamous cell
carcinoma of the esophagus. J Gastroenterol Hepatol. 25:314–318.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi M, Kawachi H, Takizawa T, et al:
p53 mutation analysis of low-grade dysplasia and high-grade
dysplasia/carcinoma in situ of the esophagus using laser capture
microdissection. Oncology. 71:237–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tao DM, Xu YZ, Gu YK, et al: A study of
the carcinogenesis time and incidence of carcinoma in 46,161 cases
with normal and hyperplastic esophageal epithelia. Cancer Res Prev
Treat. 24:155–156. 1997.(In Chinese).
|
|
7
|
Sugimachi K, Sumiyoshi K, Nozoe T, Yasuda
M, Watanabe M, Kitamura K, Tsutsui S, Mori M and Kuwano H:
Carcinogenesis and histogenesis of esophageal carcinoma. Cancer.
75(Suppl): 1440–1445. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kamangar F, Diaw L, Wei WQ, et al: Serum
pepsinogens and risk of esophageal squamous dysplasia. Int J
Cancer. 124:456–460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen W, Abnet CC, Wei WQ, Roth MJ, Lu N,
Taylor PR, Pan QJ, Luo XM, Dawsey SM and Qiao YL: Serum markers as
predictors of esophageal squamous dysplasia and early cancer.
Anticancer Res. 24:3245–3249. 2004.PubMed/NCBI
|
|
10
|
Shamma A, Doki Y, Shiozaki H, Tsujinaka T,
Yamamoto M, Inoue M, Yano M and Monden M: Cyclin D1 overexpression
in esophageal dysplasia: a possible biomarker for carcinogenesis of
esophageal squamous cell carcinoma. Int J Oncol. 16:261–266.
2000.PubMed/NCBI
|
|
11
|
Wang WC, Wu TT, Chandan VS, Lohse CM and
Zhang L: Ki-67 and ProExC are useful immunohistochemical markers in
esophageal squamous intraepithelial neoplasia. Hum Pathol.
42:1430–1437. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sakamoto K, Fujii T, Kawachi H, Miki Y,
Omura K, Morita K, Kayamori K, Katsube K and Yamaguchi A: Reduction
of NOTCH1 expression pertains to maturation abnormalities of
keratinocytes in squamous neoplasms. Lab Invest. 92:688–702. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
ENCODE Project Consortium. Birney E,
Stamatoyannopoulos JA and Dutta A: Identification and analysis of
functional elements in 1% of the human genome by the ENCODE pilot
project. Nature. 447:799–816. 2007.
|
|
14
|
Kurokawa R, Rosenfeld MG and Glass CK:
Transcriptional regulation through noncoding RNAs and epigenetic
modifications. RNA Biol. 6:233–236. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moran VA, Perera RJ and Khalil AM:
Emerging functional and mechanistic paradigms of mammalian long
non-coding RNAs. Nucleic Acids Res. 40:6391–6400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kugel JF and Goodrich JA: Non-coding RNAs:
key regulators of mammalian transcription. Trends Biochem Sci.
37:144–151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Costa FF: Non-coding RNAs: Meet thy
masters. BioEssays. 32:599–608. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar
|
|
19
|
Chen ZL, Zhao XH, Wang JW, et al:
microRNA-92a promotes lymph node metastasis of human esophageal
squamous cell carcinoma via E-cadherin. J Biol Chem.
286:10725–10734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo Y, Chen Z, Zhang L, et al: Distinctive
microRNA profiles relating to patient survival in esophageal
squamous cell carcinoma. Cancer Res. 68:26–33. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
IAkagi I, Miyashita M, Ishibashi O,
Mishima T, Kikuchi K, Makino H, Nomura T, Hagiwara N, Uchida E and
Takizawa T: Relationship between altered expression levels of
MIR21, MIR143, MIR145, and MIR205 and clinicopathologic features of
esophageal squamous cell carcinoma. Dis Esophagus. 24:523–530.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hong L, Han Y, Zhang H, Li M, Gong T, Sun
L, Wu K, Zhao Q and Fan D: The prognostic and chemotherapeutic
value of miR-296 in esophageal squamous cell carcinoma. Ann Surg.
251:1056–1063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tian D, Sun S and Lee JT: The long
noncoding RNA, Jpx, is a molecular switch for X chromosome
inactivation. Cell. 143:390–403. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kretz M, Siprashvili Z, Chu C, et al:
Control of somatic tissue differentiation by the long non-coding
RNA TINCR. Nature. 493:231–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kretz M, Webster DE, Flockhart RJ, et al:
Suppression of progenitor differentiation requires the long
noncoding RNA ANCR. Gene Dev. 26:338–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pang KC, Dinger ME, Mercer TR, et al:
Genome-wide identification of long noncoding RNAs in
CD8+T cells. J Immunol. 182:7738–7748. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aoki K, Harashima A, Sano M, Yokoi T,
Nakamura S, Kibata M and Hirose T: A thymus-specific noncoding RNA,
Thy-ncR1, is a cytoplasmic riboregulator of MFAP4 mRNA in immature
T-cell lines. BMC Mol Bio. 11:992010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khaitan D, Dinger ME, Mazar J, et al: The
melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates
apoptosis and invasion. Cancer Res. 71:3852–3862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Du Y, Kong G, You X, et al: Elevation of
highly up-regulated in liver cancer (HULC) by hepatitis B virus X
protein promotes hepatoma cell proliferation via down-regulating
p18. J Biol Chem. 287:26302–26311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin G, Sun J, Isaacs SD, et al: Human
polymorphisms at long non-coding RNAs (lncRNAs) and association
with prostate cancer risk. Carcinogenesis. 32:1655–1659. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gibb EA, Enfield KS, Stewart GL, Lonergan
KM, Chari R, Ng RT, Zhang L, MacAulay CE, Rosin MP and Lam WL: Long
non-coding RNAs are expressed in oral mucosa and altered in oral
premalignant lesions. Oral Oncol. 47:1055–1061. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mercer TR, Dinger ME, Mariani J, et al:
Noncoding RNAs in long-term memory formation. Neuroscientist.
14:434–445. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huo JS and Zambidis ET: Pivots of
pluripotency: the roles of non-coding RNA in regulating embryonic
and induced pluripotent stem cells. Biochim Biophys Acta.
1830.2385–2394. 2013.PubMed/NCBI
|
|
34
|
Ota T, Suzuki Y, Nishikawa T, et al:
Complete sequencing and characterization of 21,243 full-length
human cDNAs. Nat Genet. 36:40–45. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun J, Zhou M, Mao ZT, et al: Systematic
analysis of genomic organization and structure of long non-coding
RNAs in the human genome. FEBS Lett. 587:976–982. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Okazaki Y, Furuno M, Kasukawa T, et al;
FANTOM Consortium; RIKEN Genome Exploration Research Group Phase
I&II Team. Analysis of the mouse transcriptome based on
functional annotation of 60,770 full-length cDNAs. Nature.
420:563–573. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: insights into functions. Nat Rev Genet.
10:155–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen FJ, Sun M, Li SQ, et al: Upregulation
of the long non-coding RNA HOTAIR promotes esophageal squamous cell
carcinoma metastasis and poor prognosis. Mol Carcinog. 52:908–915.
2013. View Article : Google Scholar
|
|
41
|
Cao W, Wu W, Shi F, et al: Integrated
analysis of long noncoding RNA and coding RNA expression in
esophageal squamous cell carcinoma. Int J Genomics.
2013:4805342013.PubMed/NCBI
|
|
42
|
Li J, Chen Z, Tian L, et al: LncRNA
profile study reveals a three-lncRNA signature associated with the
survival of patients with oesophageal squamous cell carcinoma. Gut.
Feb 12–2014.(Epub ahead of print). View Article : Google Scholar
|
|
43
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li SQ, Chen FJ and Cao XF: Distinctive
microRNAs in esophageal tumor: early diagnosis, prognosis judgment,
and tumor treatment. Dis Esophagus. 26:288–298. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ørom UA, Derrien T, Beringer M, et al:
Long noncoding RNAs with enhancer-like function in human cells.
Cell. 143:46–58. 2010.
|
|
46
|
Wamstad JA, Alexander JM, Truty RM, et al:
Dynamic and coordinated epigenetic regulation of developmental
transitions in the cardiac lineage. Cell. 151:206–220. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vallot C, Huret C, Lesecque Y, Resch A,
Oudrhiri N, Bennaceur-Griscelli A, Duret L and Rougeulle C: XACT, a
long noncoding transcript coating the active X chromosome in human
pluripotent cells. Nat Genet. 45:239–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Redrup L, Branco MR, Perdeaux ER, et al:
The long noncoding RNA Kcnq1ot1 organises a lineage-specific
nuclear domain for epigenetic gene silencing. Development.
136:525–530. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pasmant E, Sabbagh A, Vidaud M and Bièche
I: ANRIL, a long, noncoding RNA, is an unexpected major hotspot in
GWAS. FASEB J. 25:444–448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Braconi C, Kogure T, Valeri N, Huang N,
Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM and Patel T:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y,
Chen N, Sun F and Fan Q: CREB up-regulates long non-coding RNA,
HULC expression through interaction with microRNA-372 in liver
cancer. Nucleic Acids Res. 38:5366–5383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Roth C, Stückrath I, Pantel K, et al: Low
levels of cell-free circulating miR-361-3p and miR-625*
as blood-based markers for discriminating malignant from benign
lung tumors. PloS One. 7:e382482012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tanaka H, Nakashiro KI, Oka R, et al:
MicroRNA-361-3p functions as an oncogenic microRNA in human oral
cancer cells. Cancer Res. 71(Suppl 1): 1452011. View Article : Google Scholar
|
|
54
|
Hughes AH, Sharma G, Roy S, et al:
microRNA expression and mRNA transcripts in clear cell
(conventional) renal cell carcinoma. Lab Invest. 91(Suppl 1s):
199A–200A. 2011.
|
|
55
|
Xiong W, Chen X, Liu F, et al: MiRNA
expression profile of microvesicles derived from K562 cells. Blood
(ASH Annual Meeting Abstracts). 118:44112011.
|
|
56
|
Sultan A, Kiet T, Chan J, et al:
Significance of microRNAs in determining taxane resistance in
ovarian cancer. Gynecol Oncol. 125(Suppl 1): S1312012. View Article : Google Scholar
|
|
57
|
Miyata H, Doki Y, Shiozaki H, et al:
CDC25B and p53 are independently implicated in radiation
sensitivity for human esophageal cancers. Clin Cancer Res.
6:4859–4865. 2000.PubMed/NCBI
|
|
58
|
Xue LY, Hu N, Song YM, et al: Tissue
microarray analysis reveals a tight correlation between protein
expression pattern and progression of esophageal squamous cell
carcinoma. BMC Cancer. 6:2962006. View Article : Google Scholar
|
|
59
|
Li JM, Wang J, Liu H, et al: The
expressions of CDC25A and CDC25B in esophageal squamous cell
carcinoma and their relationship with cell apoptosis and
proliferation. Tumor. 28:586–590. 2008.(In Chinese).
|
|
60
|
Jiang Y, Liang ZD, Wu TT, et al:
Ataxia-telangiectasia mutated expression is associated with tobacco
smoke exposure in esophageal cancer tissues and benzo[a]pyrene diol
epoxide in cell lines. Int J Cancer. 120:91–95. 2007.PubMed/NCBI
|
|
61
|
Wan Y and Wu XY: Expression and clinical
significance of DAPK1 and CD147 in esophageal squamous cell
carcinoma. Zhonghua Zhong Liu Za Zhi. 34:44–48. 2012.(In
Chinese).
|
|
62
|
Daigeler A, Chromik AM, Geisler A, et al:
Synergistic apoptotic effects of taurolidine and TRAIL on squamous
carcinoma cells of the esophagus. Int J Oncol. 32:1205–1220. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fukuda K, Sakakura C, Miyagawa K, et al:
Differential gene expression profiles of radioresistant oesophageal
cancer cell lines established by continuous fractionated
irradiation. Br J Cancer. 91:1543–1550. 2004. View Article : Google Scholar
|