Introduction
Acute lung injury (ALI) is a clinical syndrome
characterized by pulmonary edema and associated with a high
mortality rate (1). The
pathogenesis of ALI is poorly understood; however, clinical studies
have demonstrated that the regulation of epithelial sodium channel
(ENaC)-mediated alveolar fluid clearance may represent an effective
treatment strategy to improve the outcome for patients with ALI
(2,3). ENaC is composed of three homologous
subunits, α, β and γ (4). The α
subunit is essential for the functional transport of Na+
and H2O out of the airway lumen. The physiological
importance of α-ENaC in the lung has been demonstrated in a study
of α-ENaC-knockout mice, where respiratory distress and mortality
were observed ≤40 h after birth, as a consequence of an inability
to clear fluid from the lungs (5).
Furthermore, experimental evidence has indicated that a reduction
in α-ENaC expression may impair the resolution of pulmonary edema
in patients with ALI (6).
To investigate the molecular mechanisms associated
with ALI, a variety of experimental models have been used. The
induction of lung injury using intra-tracheal administration of
lipopolysaccharide (LPS) has represented a useful model for
studying ALI, as it avoids multi-organ failure (7). LPS is a prototypical endotoxin that
is a key component of the outer membrane of gram-negative bacteria,
including Pseudomonas aeruginosa. LPS has been demonstrated
to modify Na+ transport in the airway epithelium by
regulating either ENaC mRNA expression (8) or the ENaC channel current (9). The effect of LPS on α-ENaC expression
in rats and airway cell lines is controversial, as expression has
been observed to increase and decrease (10,11),
and in certain studies, to remain unchanged following LPS induction
(9,12).
Endotoxin-induced inflammation has been observed to
affect ALI, and ENaC channels have been identified to have a
significant role in the reabsorption of edema fluid; therefore, an
understanding of the impact of endotoxins on ENaC regulation may be
of major significance. The present study aimed to analyze the
regulation of α-ENaC expression in LPS models of ALI in
vitro and in vivo.
Materials and methods
Materials
LPSs from Escherichia coli (serotype, 055:B5)
were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rabbit
anti-α-ENaC monoclonal antibodies, horseradish peroxidase
(HRP)-labeled goat anti-rabbit immunoglobulin G (IgG) and TRIzol
were purchased from Invitrogen Life Technologies (Carlsbad, CA,
USA). A PrimeScript RT Reagent kit with gDNA Eraser and SYBR Green
Premix Ex Taq were obtained from Takara Bio, Inc. (Tokyo, Japan).
Other materials and reagents were purchased from Beyotime Co.
(Shanghai, China).
Animals and LPS treatment
Male and female Chinese Kun Ming mice, aged 6–7
weeks and weighing 18–22 g, were purchased from Guangdong
Experimental Animal Center (Guangzhou, China). Mice were maintained
in a temperature- and humidity-controlled room, with a 12-h
dark/light cycle, and fed on a standard laboratory diet with water.
All experimental procedures were approved by the Animal Care and
Use Committee of the School of Life Sciences (Sun Yat-Sen
University, Guangzhou, China).
Following adjustment to their environment, the mice
were randomly divided into three groups of 12 as follows: A naive
group as the control, the LPS 8-h group and the LPS 24-h group. The
mice were anesthetized using an intraperitoneal injection of 3.5%
chloral hydrate and fixed on a board at an angle of 50° in the
supine position. A total of 50 μl phosphate-buffered saline (PBS)
containing 40 μg LPS was instilled into the trachea of the mice in
the LPS 8-h and 24-h groups, using a microliter injector. The mice
in the control group were instilled with 50 μl PBS alone. Following
intratracheal instillation, the mice were placed in a vertical
position and spun for 0.5 min to ensure even distribution of the
instillation throughout the lungs (13). The mice were sacrificed at 8 and 24
h post-LPS instillation, respectively. Pathological findings, the
lung wet-to-dry weight (W/D) ratio and ENaC mRNA expression were
then evaluated.
Lung W/D ratio
The mice were sacrificed by heart bloodletting using
vacuum tubes from the left side of heart at 8 and 24 h post-LPS
instillation, respectively. The whole lungs of six mice were
removed and weighed prior to being placed in an oven at 80°C for 48
h to obtain the dry weight. The lung W/D ratio was calculated to
assess tissue edema.
Lung histological analysis
Following sacrifice, the right lungs from six mice
were fixed in 10% formalin, embedded in paraffin and cut into
3–5-μm sections for histopathological analysis. The left lungs were
stored at −80°C for RNA extraction. Hematoxylin and eosin (H&E)
staining was performed in accordance with standard methods. Slides
(n=6) were analyzed using light microscopy by two blinded
observers, and the lung tissue damage was graded on a scale of 0
(best) to 4 (worst) in accordance with combined assessments of
alveolar congestion, edema, neutrophil infiltration, atelectasis
and necrosis. The total lung injury score was calculated by adding
the average scores for each individual based on the severity of the
injury.
Immunohistochemistry of α-ENaC
expression
Tissue sections were deparaffinized and rehydrated
for immunohistochemistry. Samples were treated with All-Purpose
Powerful Antigen Retrieval Solution (Beyotime Co.) at 95°C, prior
to being blocked at room temperature using 5% bovine serum albumin
(BSA; Invitrogen Life Technologies) and incubated with rabbit
anti-α-ENaC monoclonal antibodies (1:200 in PBS with 2% BSA).
Following washing, the sections were incubated with HRP-labeled
goat anti-rabbit IgG (1:500) for 30 min at room temperature. The
HRP-labeled reagents were detected using a DAB Horseradish
Peroxidase Color Development kit (Beyotime Co.). Brown staining in
the airway and alveolar epithelial cells was considered to indicate
a positive result for α-ENaC expression. Results were evaluated
semi-quantitatively according to optical density values of positive
expression using the Medical Image Analysis System, HMIAS-2000
(Qianping Image Co., Wuhan, China).
Quantitative polymerase chain reaction
(qPCR) for analysis of α-ENaC mRNA expression in lung tissues
Total RNA was extracted from 50 mg lung tissue using
TRIzol reagent, in accordance with the manufacturer’s instructions.
The reverse transcription reaction was performed using the
PrimeScript RT Reagent kit with gDNA Eraser. To quantitatively
determine the levels of α-ENaC mRNA expression, qPCR analysis was
performed in the Roche LightCycle 480 System (Roche, Mannheim,
Germany) using SYBR Green Premix Ex Taq and the following cycle
conditions: 95°C for 30 sec, followed by 40 cycles of 95°C for 10
sec, 62°C for 20 sec and 72°C for 30 sec). The identity and purity
of the PCR products were assessed using a melting curve analysis.
α-ENaC mRNA expression was quantified using a comparative cycle
threshold method and was normalized using GAPDH as an endogenous
control. The primer sequences used for qPCR analysis were
synthesized by Invitrogen Life Technologies (Guangzhou, China) and
were as follows: Mouse α-ENaC, 5′-CACCTTTGCTTTTGTGAACTCG-3′
(forward) and 5′-CATCCCTGAGCACAGTTCAGTC-3′ (reverse); mouse GAPDH,
5′-ACCCAGAAGACTGTGGATGG-3′ (forward) and 5′-CACATTGGGGGTAGGAACAC-3′
(reverse).
Cell culture and measurement of α-ENaC
mRNA expression in A549 cells
The human lung alveolar epithelial type II A549 cell
line was purchased from the American Type Culture Collection
(Rockville, MD, USA) and maintained in RPMI-1640 medium
supplemented with 10% fetal calf serum and 1%
penicillin/streptomycin in a humidified incubator with 95% air and
5% CO2 at 37°C until the cells reached confluence.
Confluent A549-monolayers (5×105 cells)
were grown in six-well plates (Costar; Corning Inc., Corning, NY,
USA) for 24 h. The cells were starved for 24 h with RPMI-1640
containing 1% fetal bovine serum prior to LPS treatment. LPS was
suspended in culture medium and used at a final concentration of 10
μg/ml. Following exposure to LPS for 1, 3, 8, 24 and 48 h, total
cellular RNA was extracted from the A549 cells using TRIzol
reagent. α-ENaC mRNA expression was then measured using qPCR
analysis as aforementioned. The primer sequences were as follows:
Human α-ENaC, 5′-TTTCACCAAGTGCCGGAAG-3′ (forward) and
5′-GCCATCGTGAGTAACCAGCA-3′ (reverse); human GAPDH,
5′-GAAGGTGAAGGTCGGAGTC-3′ (forward) and 5′-GAAGATGGTGATGGGATTTC-3′
(reverse). Prior to the study, an MTT reduction assay was used to
confirm that this concentration of LPS (10 μg/ml) had no effect on
A549 cell viability within 48 h.
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analyses were performed using SPSS
statistical software 16.0 (SPSS, Inc., Chicago, IL, USA). A one-way
analysis of variance, followed by the Student-Newman-Keuls test
were used for comparing the treatment results. P<0.05 was
considered to indicate a statistically significant difference.
Results
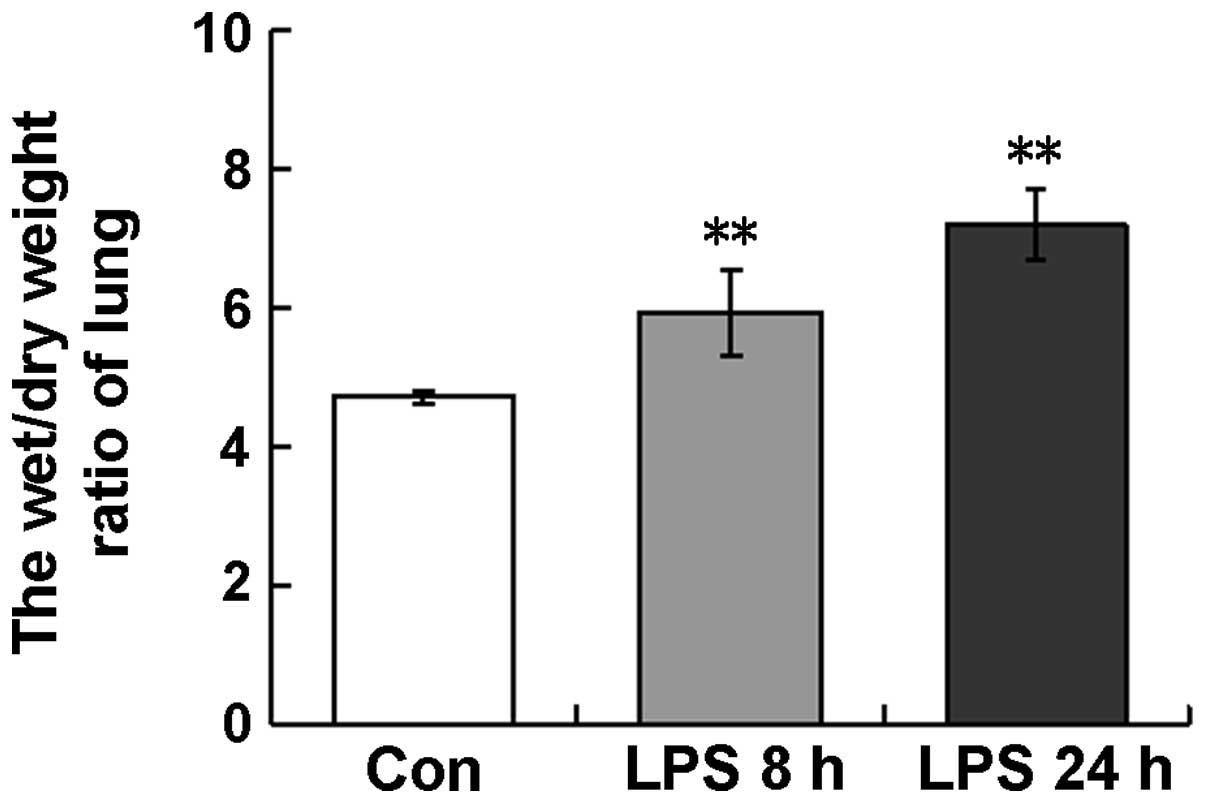
Lung W/D ratio
The W/D ratio is frequently used as an index of
pulmonary edema. In the ALI model used in the present study, the
lung W/D ratios in the LPS-treated mice were 5.9±0.6 and 6.2±0.5 at
8 and 24 h, respectively, which were significantly higher than that
in the control mice (4.7±0.1) (Fig.
1). This observation indicated that LPS induced the development
of the pulmonary edema.
Pathological findings and
immunohistochemistry of α-ENaC
Histopathological examinations were performed using
H&E staining and light microscopy (Fig. 2). Alveolar congestion, edema,
neutrophil infiltration, atelectasis and necrosis were semi-scored
by the blinded observers (Table
I). At 8 h post-LPS instillation (Fig. 2B), compared with the control,
marked pathological alterations were detected, including
infiltration of inflammatory cells into the alveolar space,
atelectasis, necrosis and interstitial and alveolar edema (Fig. 2A). The histological damage observed
at 8 h was relatively mild compared with that at 24 h (Fig. 2C), where alveolar congestion,
alveolar atelectasis and fusion, and increased septal thickness as
a consequence of inflammatory cell infiltration, were observed.
 | Figure 2LPS-induced lung morphology,
immunohistochemistry of α-ENaC and their semiquantitative scores.
(A–C) Morphological changes in the lung detected using H&E
staining. Control KM mice that were instilled with 50 μl PBS
exhibited no specific ALI-associated changes in lung morphology. In
the ALI models, 50 μl PBS containing 40 μg LPS was instilled into
the trachea of the KM mice. Compared with (A) the control,
significant pathological changes, including inflammation, edema and
interalveolar septum thickening, were observed in (B) the
LPS-treated 8-h group. (C) At 24 h, the pathological changes were
more severe, including interstitial and intra-alveolar hemorrhage
and alveolus atelectasis and fusion. (D–F) Immunohistochemistry of
the α-ENaC channel. Compared with (D) the control, the
immunoreactivity of α-ENaC was stronger in (E) the LPS-treated 8-h
group and reduced in (F) the 24-h group. (G–H) The
semi-quantitative scores of (G) histopathology and (H)
immunohistochemistry. Each bar represents the mean ± standard
deviation (**P<0.01 vs. control group, n=6). LPS,
lipopolysaccharide; ENaC, epithelial sodium channel; ALI, acute
lung injury; PBS, phosphate-buffered saline; H&E, hematoxylin
and eosin; KM, Chinese Kun Ming; Con, control. |
 | Table ISemi-score analysis of
morphopathological changes in the lung tissues of KM mice. |
Table I
Semi-score analysis of
morphopathological changes in the lung tissues of KM mice.
| Group | Con | LPS 8 h | LPS 24 h |
|---|
| Alveolar
congestion | 1.0 | 1.5 | 3.0 |
| Edema | 0.5 | 2.0 | 3.5 |
| Neutrophil
infiltration | 0.5 | 2.5 | 3.5 |
| Atelectasis | 0.5 | 2.0 | 4.0 |
| Necrosis | 0.0 | 2.0 | 3.5 |
As shown in Fig.
2D–F, significant α-ENaC expression was observed at the apical
side of the airway and alveolar epithelial cells, represented by
the strong brown staining. Compared with the control (Fig. 2D), the immunoreactivity of α-ENaC
was observed to increase at 8 h post-LPS treatment (Fig. 2E) and decrease by 24 h (Fig. 2F). This finding indicated that
α-ENaC protein expression increased 8 h after LPS treatment, but
declined with the development of the pulmonary edema.
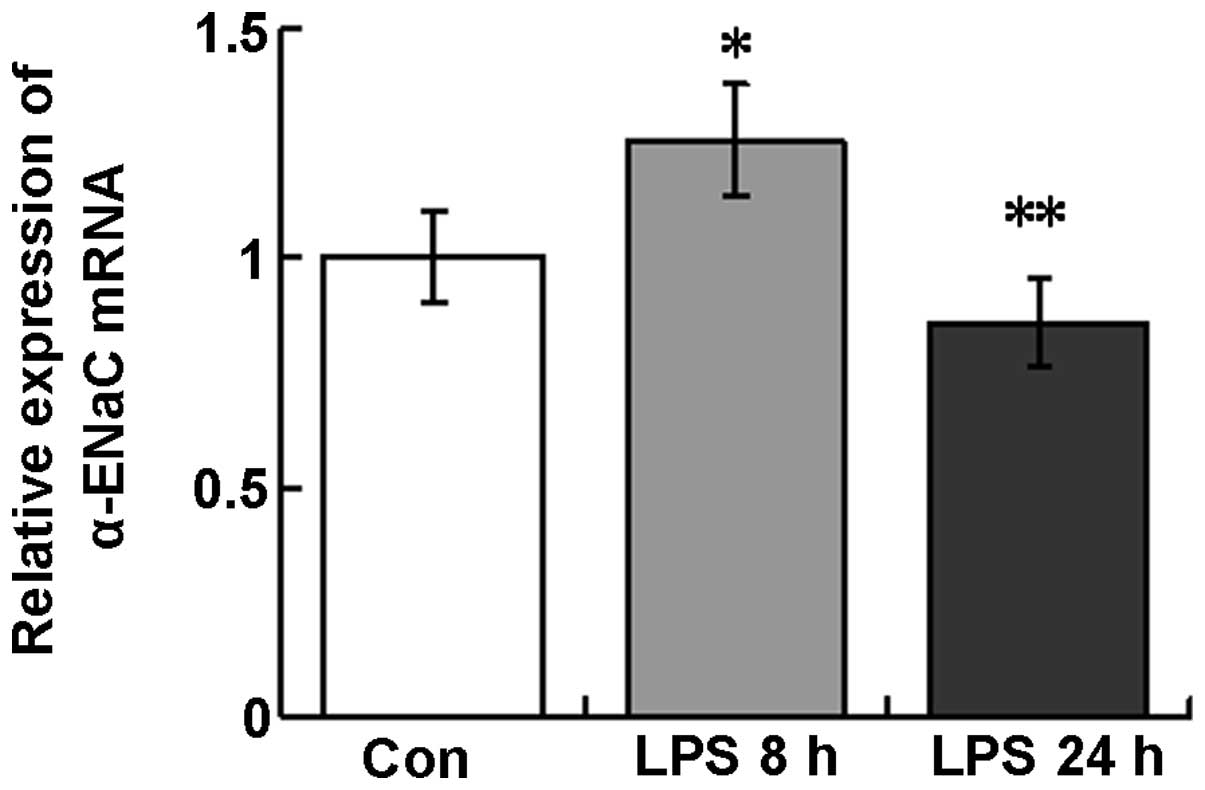
α-ENaC mRNA expression in lung
tissues
Following LPS instillation, α-ENaC mRNA expression
was observed to increase at 8 h (120.7±22.1%) and decrease at 24 h
(85.9±14.6%) in the tissues of the whole lungs compared with those
of the control (Fig. 3).
Therefore, α-ENaC mRNA and protein demonstrate similar temporal
expression patterns in response to LPS treatment in mouse lung
tissues.
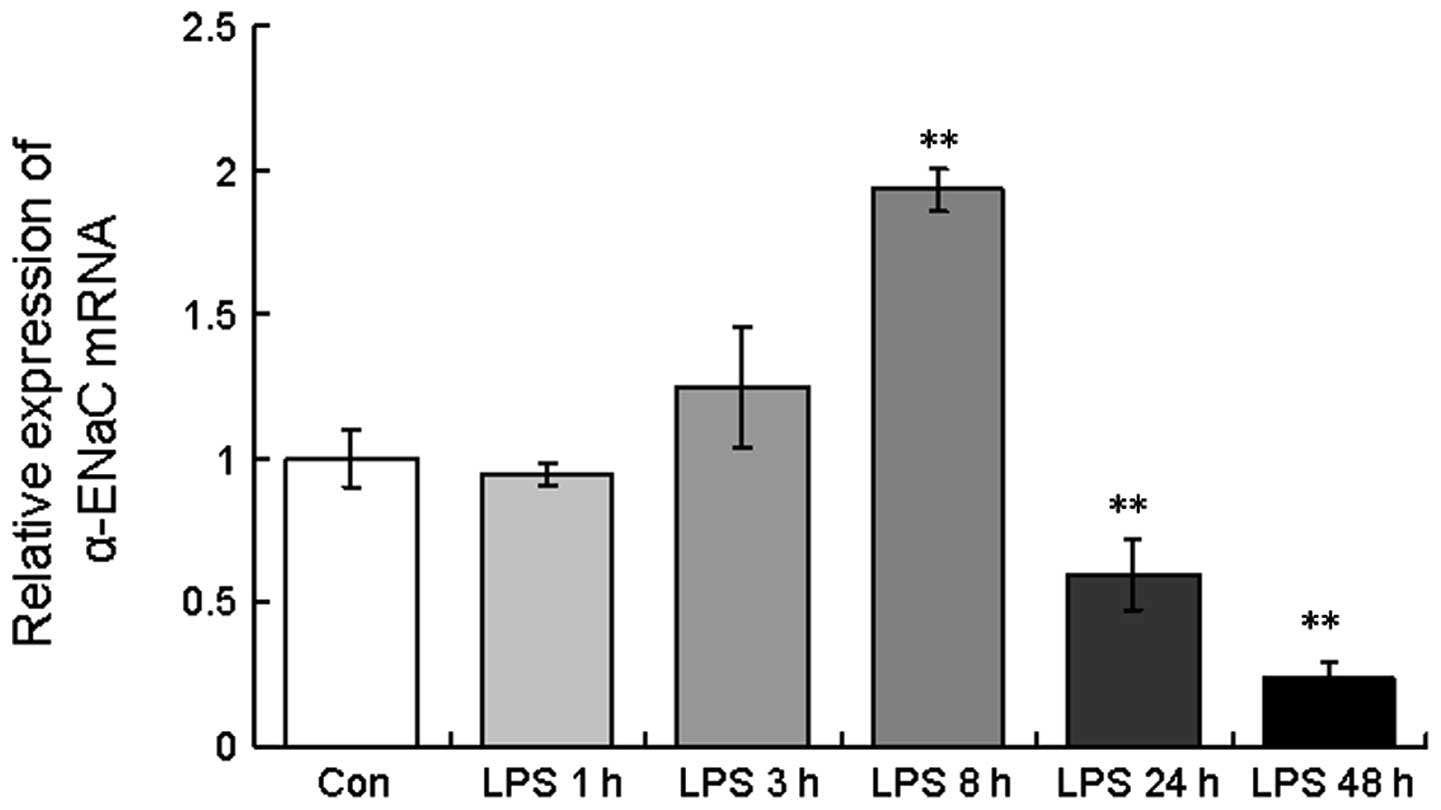
mRNA expression of α-ENaC in A549
cells
As shown in Fig. 4,
the level of α-ENaC mRNA was highly modulated in the A549 cells
with respect to the different LPS exposure times, with an increase
in α-ENaC mRNA expression observed at 3 and 8 h post-LPS treatment
(124.6±20.8 and 193.3±7.5%, respectively). However, following
continuous LPS treatment for 24 and 48 h, α-ENaC mRNA expression
decreased (59.6±12.3 and 23.6±5.5%, respectively; Fig. 4), consistent with the in
vivo findings.
Discussion
The present study analyzed the regulation of α-ENaC
expression in LPS models of ALI at different pathological stages
in vitro and in vivo. An increase in α-ENaC
expression was observed at 8 h post-treatment, which decreased
thereafter. This demonstrated that the modulation of α-ENaC by LPS
may be biphasic, with a transient increase in the early stage of
ALI followed by a sustained decrease thereafter.
Numerous previous studies have reported that ENaC
may be regulated by LPS; however, these findings are discrepant, as
both increases and decreases in expression were observed (10,11).
Moreover, it has also been reported that ENaC expression was not
affected by LPS induction (9,12).
In human H441 airway epithelial cells, α-ENaC mRNA and protein
levels have been observed to be downregulated by LPS (10). In addition, in an LPS-induced mouse
model of middle ear mucosa inflammation, the level of α-ENaC
expression was found to decrease in the initial 12-h period,
normalize at 24 h and then increase thereafter (11). However, studies by Dodrill and
Fedan (9) and Dodril et al
(12) reported that systemic
administration of LPS was capable of increasing the activity of the
Na+ channel, but with no impact on ENaC transcription.
Only a single previous study is consistent with the results of the
present study; this previous study demonstrated that following
infection with Pseudomonas aeruginosa, a bacterium
frequently present in patients with bronchiectasis, ENaC expression
in the lung was increased over the initial 24 h, but was followed
by a sustained decrease on days three and seven (8). The findings of the present study
combined with those of previous reports indicate that the mechanism
by which LPS modulates ENaC expression is complex. It was
hypothesized in the present study that α-ENaC may not be a direct
genetic marker associated with LPS exposure, but a general response
of the lung to the pathological changes involved in the development
of ALI. The degree of lung injury varies depending on the dose of
LPS and the duration of exposure, which may account for the varying
effects observed for LPS on ENaC expression.
The results of the present study showed that α-ENaC
expression increased in the early stages of ALI, which may
represent a self-repair mechanism induced by the body. In this
stage, the pathological changes were relatively mild; however, lung
interstitial and mild alveolar edema were observed, along with an
increase in Na+ transport and alveolar liquid clearance,
as an attempt to maintain dry alveolar spaces. It has been reported
that in mild-to-moderate lung injury, Na+ transport may
be upregulated by stress hormones or by catecholamine-dependent
mechanisms (14). However, in the
late stage of ALI, inflammatory cytokines induced by LPS are
produced in excess, and are capable of promoting a cascade of
deleterious events resulting in endothelial and epithelial
dysfunction, which may lead to a decrease in the expression of
ENaC. Furthermore, numerous cytokines, including interleukin
(IL)-1β (15), IL-4 (16), interferon-γ (17) and transforming growth factor-β1
(18), have been reported to be
involved in the regulation of ENaC expression. Moreover, it has
been shown that the impaired gas exchange associated with the
development of pulmonary edema may cause severe tissue hypoxia, and
hypoxia has been reported to impair alveolar edema clearance
through mechanisms that downregulate the expression and activity of
ENaC (19,20). Therefore, it may be possible that
the inflammation, hypoxia and endothelial and epithelial damage
associated with severe edema downregulate the expression of α-ENaC
and attenuate alveolar edema clearance.
Notably, in the present study, the biphasic
modulation of α-ENaC expression, with an increase at 8 h followed
by a decrease thereafter, may explain the contradictory reports
concerning ENaC expression following endotoxin or bacteria
infection in the lung. Instillation of endotoxin into rat lungs and
acute bacterial pneumonia in rats has been found to upregulate
sodium transport and increase alveolar epithelial fluid clearance
(21,22). However, in late pneumonia or severe
ALI with pulmonary edema, a significant decrease in α-ENaC
expression has been identified, which is associated with a reduced
ability for lung fluid clearance (22). The modulation of α-ENaC expression
reported in the present study, may explain these contradictory
data. Understanding the mechanisms responsible for the early
stimulation and late inhibition of ENaC expression may be of
clinical significance to improve the outcome for patients with
endotoxin-induced ALI.
In conclusion, the present study has indicated that
LPS may modulate α-ENaC expression in a biphasic manner, with a
transient increase in the early stage followed by a sustained
decrease thereafter. The results of this study, in combination with
those of previous studies, indicate that LPS may modulate ENaC
expression through the induction of changes in the inflammatory
milieu or through pathological changes, including hypoxia or
endothelial and epithelial damage, rather than through a direct
mechanism. Therefore, LPS-induced α-ENaC mRNA modulation is likely
to be complex and involve mechanisms that are specific to the cell
insult. Further research is required to elucidate the specific
pathway by which bacterial LPS regulates ENaC expression.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81173475).
References
|
1
|
Maybauer MO, Maybauer DM and Herndon DN:
Incidence and outcomes of acute lung injury. N Engl J Med.
354:416–417. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ware LB and Matthay MA: Alveolar fluid
clearance is impaired in the majority of patients with acute lung
injury and the acute respiratory distress syndrome. Am J Respir
Crit Care Med. 163:1376–1383. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berthiaume Y, Folkesson HG and Matthay MA:
Lung edema clearance: 20 years of progress: invited review:
alveolar edema fluid clearance in the injured lung. J Appl Physiol
(1985). 93:2207–2213. 2002.PubMed/NCBI
|
|
4
|
Canessa CM, Schild L, Buell G, et al:
Amiloride-sensitive epithelial Na+ channel is made of
three homologous subunits. Nature. 367:463–467. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hummler E, Barker P, Gatzy J, et al: Early
death due to defective neonatal lung liquid clearance in
alpha-ENaC-deficient mice. Nat Genet. 12:325–328. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Egli M, Duplain H, Lepori M, et al:
Defective respiratory amiloride-sensitive sodium transport
predisposes to pulmonary oedema and delays its resolution in mice.
J Physiol. 560:857–865. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rittirsch D, Flierl MA, Day DE, et al:
Acute lung injury induced by lipopolysaccharide is independent of
complement activation. J Immunol. 180:7664–7672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dagenais A, Gosselin D, Guilbault C, et
al: Modulation of epithelial sodium channel (ENaC) expression in
mouse lung infected with Pseudomonas aeruginosa. Respir Res.
6:22005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dodrill MW and Fedan JS:
Lipopolysaccharide hyperpolarizes guinea pig airway epithelium by
increasing the activities of the epithelial Na(+) channel and the
Na(+)-K(+) pump. Am J Physiol Lung Cell Mol Physiol. 299:L550–L558.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baines DL, Albert AP, Hazell MJ, et al:
Lipopolysaccharide modifies amiloride-sensitive Na+
transport processes across human airway cells: role of
mitogen-activated protein kinases ERK 1/2 and 5. Pflugers Arch.
459:451–463. 2010.PubMed/NCBI
|
|
11
|
Song JJ, Kwon SK, Cho CG, et al:
Expression of ENaC in LPS-induced inflammation of middle ear
mucosa. Acta Otolaryngol. 132:1145–1150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dodrill MW, Beezhold DH, Meighan T, et al:
Lipopolysaccharide increases Na(+), K(+)-pump, but not ENaC,
expression in guinea-pig airway epithelium. Eur J Pharmacol.
651:176–186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su X, Wang L, Song Y and Bai C: Inhibition
of inflammatory responses by ambroxol, a mucolytic agent, in a
murine model of acute lung injury induced by lipopolysaccharide.
Intensive Care Med. 30:133–140. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berthiaume Y and Matthay MA: Alveolar
edema fluid clearance and acute lung injury. Respir Physiol
Neurobiol. 159:350–359. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roux J, Kawakatsu H, Gartland B, et al:
Interleukin-1beta decreases expression of the epithelial sodium
channel alpha-subunit in alveolar epithelial cells via a p38
MAPK-dependent signaling pathway. J Biol Chem. 280:18579–18589.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Galietta LJ, Pagesy P, Folli C, et al:
IL-4 is a potent modulator of ion transport in the human bronchial
epithelium in vitro. J Immunol. 168:839–845. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Galietta LJ, Folli C, Marchetti C, et al:
Modification of transepithelial ion transport in human cultured
bronchial epithelial cells by interferon-gamma. Am J Physiol Lung
Cell Mol Physiol. 278:L1186–L1194. 2000.PubMed/NCBI
|
|
18
|
Frank J, Roux J, Kawakatsu H, et al:
Transforming growth factor-beta1 decreases expression of the
epithelial sodium channel alphaENaC and alveolar epithelial
vectorial sodium and fluid transport via an ERK1/2-dependent
mechanism. J Biol Chem. 278:43939–43950. 2003. View Article : Google Scholar
|
|
19
|
Clerici C and Matthay MA: Hypoxia
regulates gene expression of alveolar epithelial transport
proteins. J Appl Physiol (1985). 88:1890–1896. 2000.PubMed/NCBI
|
|
20
|
Wodopia R, Ko HS, Billian J, et al:
Hypoxia decreases proteins involved in epithelial electrolyte
transport in A549 cells and rat lung. Am J Physiol Lung Cell Mol
Physiol. 279:L1110–L1119. 2000.PubMed/NCBI
|
|
21
|
Rezaiguia S, Garat C, Delclaux C, et al:
Acute bacterial pneumonia in rats increases alveolar epithelial
fluid clearance by a tumor necrosis factor-alpha-dependent
mechanism. J Clin Invest. 99:325–335. 1997. View Article : Google Scholar
|
|
22
|
Viget NB, Guery BP, Ader F, et al:
Keratinocyte growth factor protects against Pseudomonas
aeruginosa-induced lung injury. Am J Physiol Lung Cell Mol
Physiol. 279:1199–1209. 2000.
|