Introduction
Hereditary ataxia comprises a clinically and
genetically heterogeneous group of neurodegenerative disorders
(1). Clinical features include
progressive limb and gait ataxia, loss of coordination and
disturbance of speech and occulomotor control. Lesions are common
in the spinal cord, cerebellum and brain stem; thus, the pathology
is also referred to as spinocerebellar ataxia (SCA). In 1863,
Friedreich (2) made the first
report of a case of SCA that occurred in an adolescent, and since
then it has been reported that several gene mutations are
associated with various subtypes of hereditary ataxia. In this
study, a familial case of late-onset hereditary ataxia was
identified, mimicking pontocerebellar hypoplasia (PCH) in its
clinical, neuroradiological and genetic aspects, but differing in
its age of onset. At present, seven different subtypes of PHC have
been identified, which are commonly characterized by hypoplasia and
variable atrophy of cerebellum and pons, progressive microcephaly
and variable cerebral involvement. Mutations in three tRNA splicing
endonuclease subunit genes (TSEN54, TSEN2 and TSEN34) were
demonstrated to be responsible for PCH2, PCH4 and PCH5. Mutations
in the mitochondrial tRNA arginyl synthetase gene (RARS2) are
associated with PCH6. TSEN54, RARS2 and the vaccinia related kinase
1 (VRK1) are mutated in a minority of PCH1 cases. PHC7, the latest
subtype, has not yet been clarified in detail (3,4).
Case report
The proband was a 36-year-old female referred to the
Department of Family Medicine of the Sir Run Run Shaw hospital due
to a consistently unsteady gait for 6 years. The patient initially
presented with a mildly imbalanced gait and incoordination at 30
years of age. The symptoms progressed with unsteady walking and a
tendency to fall, accompanied by coughing upon drinking and slurred
speech. No symptoms of headache, vomiting or dysphagia were
reported.
A family history investigation revealed a
four-generation family history with seven affected family members,
who presented with similar symptoms of cerebellar ataxia, including
unsteady gait, coughing upon drinking and slurred speech, in their
mid-thirties and succumbed due to complications of PCH during their
fifties or sixties (Fig. 1). No
aggravation of the symptoms was found from one generation to the
next. The genetic inheritance pattern was consistent with an
autosomal dominant model.
 | Figure 1Pedigree of a Chinese family with
pontocerebellar hypoplasia. Patient 1, onset at 30, diet at 50;
patient 2, onset at 28, died at 60; patient 3, onset at 40, died at
70; patient 4, onset at 30, died at 50; patient 5, onset at 30,
died at 65; patient 6, onset at 27, died at 63; patient 7 (the
proband), onset at 36. Filled squares and circles denote affected
males and females, respectively, indicated in the figure by arrows
1–7. Normal individuals are shown as empty symbols. The proband is
shown by the indicated arrow. D1 died of cardiac problem in his
fifties, D2 died of drowning in his twenties. |
Physical examination showed a normal appearance and
stature; however, the neurological examination revealed a series of
positive indicators of cerebellar ataxia, including a wide-based
gait, difficulty walking in a straight line, hyporeflexia of the
left pharynx reflex, enhanced muscular tension and tendon reflex of
both lower extremities and positive results for the finger-to-nose
and heal-knee-tibia tests, as well as for the Romberg and Babinski
signs (tests of proprioception and pyramidal tract,
respectively).
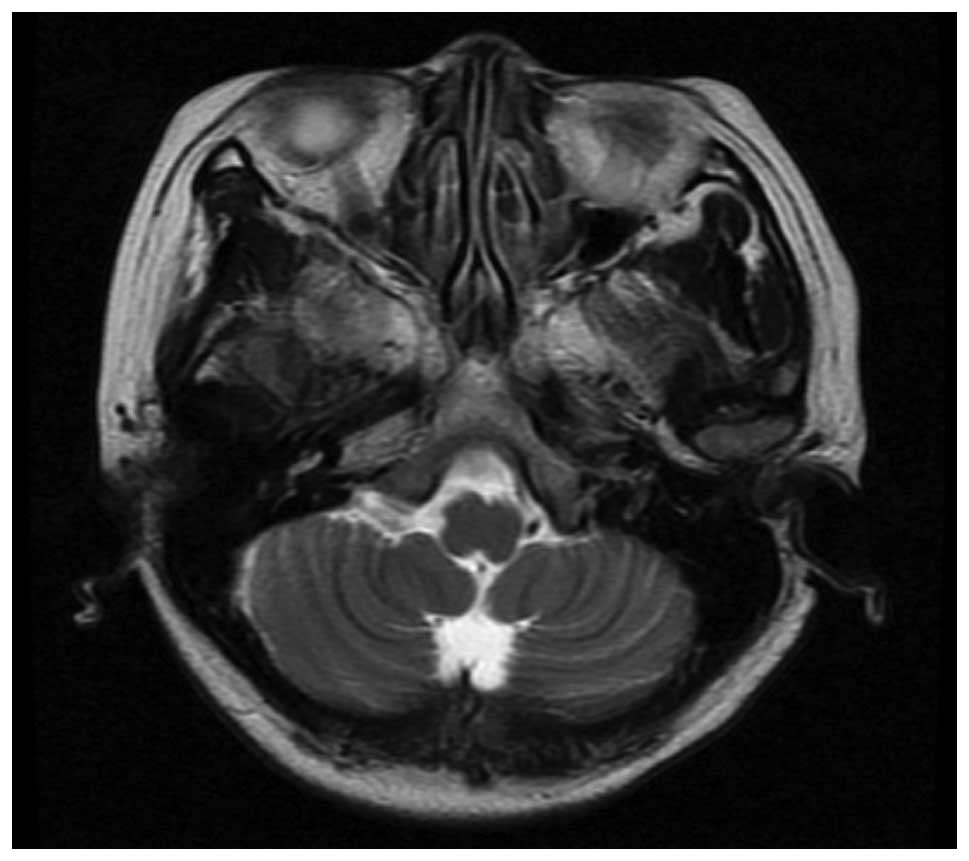
Initial laboratory investigations were normal,
although magnetic resonance imaging (MRI) of the brain identified
mild atrophy of the cerebellar hemisphere with visibly deepened
sulci (Fig. 2). From these data, a
primary diagnosis of hereditary cerebellar ataxia was
considered.
A detailed gene analysis of the patient and other
family members was conducted to determine further diagnosis and
treatment. Prior to this, ethics approval was gained from the
Ethics Committee of Zhejiang University (Hangzhou, China) and
informed consent was obtained from the patient and all family
members. Genomic DNA was extracted from nucleated blood cells of
the affected and unaffected individuals of a four-generation
Chinese family with PCH. Candidate genes were then selected for
sequencing based upon their reported involvement in PCH
development. For mutation analysis, the coding region of TSEN54 was
sequenced with DNA from one patient and other unaffected alive
family members. Other susceptible sites, including two TSEN2 sites
(c.926 A > G and c.960+1delGTAAG) and four mitochondrial
arginyl-tRNA synthetase (RARS2) sites (c.35 A > G, c.110+5 A
> G, c.1024 A > G and c.1072 C > T), were sequenced only
in the affected patient. The primers were designed according to
previous studies (5,6). Amplification was performed using a
Bori thermal cycler (Bori Technology Co., Ltd., Hangzhou, China)
and polymerase chain reaction (PCR) products were then separated by
polyacrylamide gel electrophoresis and directly sequenced using an
ABI BigDye® Terminator Cycle Sequencing kit (Applied
Biosystems, Austin, TX, USA) according to manufacturer’s
instructions and run on an ABI 3100 sequencer. Sequencing results
were analyzed using the DNASTAR® software program
package (DNASTAR, Inc., Madison, WI, USA).
A novel c.254A > T heterozygous mutation in the
coding region of TSEN54 was found by direct sequencing in the
affected family members. No other mutations were detected in any
other exon of TSEN54, TSEN2, RARS2 or vVRK1 in the affected
patient, and no mutations were found in any unaffected family
members. The mutation resulted in an amino acid substitution from
glutamic acid to valine at amino acid 85 (Fig. 3). For the differential diagnosis of
SCAs, another hereditary ataxia disorder prevalent in the Chinese
population, a CAG repeat mutation in SCA1, 2, 3, 6, 7 and 17 was
screened for by PCR and DNA sequencing techniques. A CAG repeat was
not found in any of the SCA genes in the affected patients.
Discussion
PCH comprises a group of neurodegenerative disorders
characterized by a small cerebellum and brainstem and progressive
microcephaly. The seven currently described PCH subtypes are
distinguished by additional characteristics. Two subtypes, PCH2 and
PCH4, are the most common, with dyskinesia and dystonia observed in
PCH2 (7) and more severe symptoms
and prenatal onset in PCH4. The most predominant clinical
manifestation is cerebellar ataxia. Autonomic nervous-system
disturbance and extra-pyramidal symptoms are also frequently
observed in patients with PCH. Several genes have been shown to
contribute to the manifestation and symptoms of PCH, with all genes
currently identified being involved in tRNA processing: PCH2B is
caused by a TSEN2 mutation, PCH2C is caused by TSEN34 mutations,
PCH6 is caused by mutations in RARS2 (5) and mutations in TSEN54, the gene
encoding noncatalytic subunits of the tRNA-splicing endonuclease
complex, have been detected in patients exhibiting symptoms
characteristic of PCH2 and PCH4 (6). A total of >20 mutations have been
identified within the TSEN54 gene in various subtypes of PCH, with
A307S being the most commonly occurring mutation in families with
inherited PCH (3).
The radiological features of PCH are hypoplasia and
variable atrophy of the cerebellum and the pons. By MRI, the
typical characteristic of PCH2 is a ‘dragonfly-like’ cerebellar
pattern (4), while patients with
PCH4 exhibit C-shaped inferior olives (8). The case presented in this study
showed a mild atrophy of the cerebellar hemisphere with visibly
deepened sulci; these radiological symptoms may correspond with the
milder symptoms and late onset. It is difficult to differentiate
PCH from other hereditary ataxia diseases from their clinical
manifestations. SCA is a relatively common neurological
degenerative disease with hereditary disparity. Abnormal CAG
repeats found in the coding regions of SCA1, 2, 3, 6, 7 and 17 and
DRPLA have been cloned (9). No
abnormal CAG repeats were found in any of the affected patients in
the present case.
VRK1 has been reported to be associated with a mild
presentation of PCH1 (10), and
mutations in the transfer RNA splicing endonuclease subunit genes
TSEN54, TSEN2 and TSEN34 have been associated with PCH2 and 4. In
addition, mutations in RARS2 have been identified in patients with
PCH6 (11).
TSEN54 is highly expressed in neurons of the pons,
cerebellar dentate and olivary nuclei during the second trimester
of pregnancy (12). Budde et
al (11) identified
homozygosity for an A307S mutation in the TSEN54 gene in 42/47
individuals with PCH2. In the presented case, a novel heterozygous
missense c.254 A > T mutation in the coding region of TSEN54 was
identified. The consequences of this mutation on protein
conformation and function and whether this results in hereditary
ataxia mimicking PCH require further investigation. It is therefore
important to perform a mutation analysis of known PCH-causing genes
in patients presenting with symptoms similar to PCH.
Acknowledgements
This study was supported by a grant from the Health
Bureau of Zhejiang Province, China (no. 2012KYA108).
References
|
1
|
Sumathipala DS, Abeysekera GS, Jayasekara
RW, Tallaksen CM and Dissanayake VH: Autosomal dominant hereditary
ataxia in Sri Lanka. BMC Neurol. 13:392013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedreich N: On degenerative atrophy of
the spinal dorsal columns. Arch Pathol Anat Physiol Klin Med.
26:391–419. 1863.(In German).
|
|
3
|
Namavar Y, Barth PG, Poll-The BT and Baas
F: Classification, diagnosis and potential mechanisms in
pontocerebellar hypoplasia. Orphanet J Rare Dis. 6:502011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Namavar Y, Barth PG, Kasher PR, et al:
Clinical, neuroradiological and genetic findings in pontocerebellar
hypoplasia. Brain. 134:143–156. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barth PG, Blennow G, Lenard HG, et al: The
syndrome of autosomal recessive pontocerebellar hypoplasia,
microcephaly, and extrapyramidal dyskinesia (pontocerebellar
hypoplasia type 2): compiled data from 10 pedigrees. Neurology.
45:311–317. 1995. View Article : Google Scholar
|
|
6
|
Maricich SM, Aqeeb KA, Moayedi Y, et al:
Pontocerebellar hypoplasia: review of classification and genetics,
and exclusion of several genes known to be important for cerebellar
development. J Child Neurol. 26:288–294. 2011. View Article : Google Scholar
|
|
7
|
Simonati A, Cassandrini D, Bazan D and
Santorelli FM: TSEN54 mutation in a child with pontocerebellar
hypoplasia type 1. Acta Neuropathol. 121:671–673. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barth PG, Aronica E, de Vries L, et al:
Pontocerebellar hypoplasia type 2: a neuropathological update. Acta
Neuropathol. 114:373–386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schöls L, Bauer P, Schmidt T, Schulte T
and Riess O: Autosomal dominant cerebellar ataxias: clinical
features, genetics, and pathogenesis. Lancet Neurol. 3:291–304.
2004.PubMed/NCBI
|
|
10
|
Renbaum P, Kellerman E, Jaron R, et al:
Spinal muscular atrophy with pontocerebellar hypoplasia is caused
by a mutation in the VRK1 gene. Am J Hum Genet. 85:281–289. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cassandrini D, Cilio MR, Bianchi M, et al:
Pontocerebellar hypoplasia type 6 caused by mutations in RARS2:
definition of the clinical spectrum and molecular findings in five
patients. J Inherit Metab Dis. 36:43–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Budde BS, Namavar Y, Barth PG, et al: tRNA
splicing endonuclease mutations cause pontocerebellar hypoplasia.
Nat Genet. 40:1113–1118. 2008. View
Article : Google Scholar : PubMed/NCBI
|