Introduction
Skeletal systems are maintained by continuous bone
remodeling, which is a process regulated by osteoblasts and
osteoclasts. Osteoblasts have a critical role in bone formation and
re-modification by producing stimulatory and inhibitory factors
that tightly regulate osteoclast formation and activity. The
functioning of osteoblasts is regulated by numerous factors
including hormones, growth factors and cytokines, such as tumor
necrosis factor-α (TNF-α) (1).
TNF-α is synthesized in the bone microenvironment and has been
demonstrated to exert pleiotropic effects on osteoblasts. High
levels of TNF-α have been identified in the tissue and gingival
crevicular fluid of patients with advanced periodontitis and
chronic periapical periodontitis (2–4).
Increased TNF-α levels have also been identified in primarily
infected root canals and the infected periapical tissue of patients
with pulpitis and chronic periapical periodontitis (5,6).
Therefore, the expression of TNF-α is considered to be correlated
with the progression of bone resorption in periodontal and
periapical diseases.
TNF-α has been reported to stimulate the production
of macrophage colony stimulating factor (M-CSF) in osteoblasts
(7,8), which binds to M-CSF receptor (CSF-1R)
on the surface of pre-osteoclasts. This binding stimulates
osteoclast differentiation and alveolar bone resorption (9,10).
Previously, interleukin-34 (IL-34) was identified as a novel
cytokine with similar characteristics to M-CSF (11). Although IL-34 shares no sequence
homology with M-CSF, IL-34 binds to the CSF-1R and promotes the
differentiation, proliferation and survival of osteoclasts. It was
also reported that IL-34 as well as M-CSF, in combination with
receptor activated nuclear factor-κB (NF-κB) ligand (RANKL),
promoted osteoclast differentiation and bone resorption in mouse
and human cell culture systems (12). However, whether TNF-α induces IL-34
expression in mouse osteoblasts has not yet been fully
investigated.
Nuclear factor-κB (NF-κB) is a transcription factor,
which is activated by numerous types of extracellular stimuli,
including bacterial products, oxidative stress and physical stress.
NF-κB is a multifunctional transcription factor that regulates
various gene expression involved in numerous cellular activities
(13,14). In unstimulated cells, NF-κB is
sequestered in the cytoplasm bound to nuclear factor of κ light
polypeptide gene enhancer in B-cell inhibitors (IκBs). When NF-κB
activation is stimulated, IκB is phosphorylated, ubiquitinated and
then degraded by the protease, thereby allowing translocation of
the liberated NF-κB from the cytoplasm to the nucleus (15). NF-κB is activated in numerous
inflammatory conditions. Activated nuclear NF-κB, in turn,
regulates the expression of cytokines and so mediates autocrine,
self-amplifying cycles of cytokine release (13,14).
Several studies have demonstrated that TNF-α induces NF-κB
activation in human or rat osteoblastic cells as well as
osteoclasts, which mediates the upregulation of interleukin-6
(IL-6) and intercellular adhesion molecule-1 (ICAM-1) (16,17).
However, whether NF-κB activation induced by TNF-α is involved in
IL-34 expression in mouse osteoblasts remains elusive.
In the present study, the expression of IL-34 in
TNF-α-treated mouse MC3T3-E1 osteoblastic cells was examined. The
involvement of NF-κB in TNF-α-induced IL-34 expression in
osteoblasts was also investigated.
Materials and methods
Materials
α-modified Eagle’s minimal essential medium (α-MEM),
OPTI-MEM and Lipofectamine™ reagent were purchased from Invitrogen
Life Technologies (Carlsbad, CA, USA); TNF-α was purchased from
Sigma-Aldrich (St. Louis, MO, USA); antibodies against NF-κB and
Eps were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz,
CA, USA); anti-lamin B1 monoclonal antibody was from Zymed (South
San Francisco, CA, USA) and caffeic acid phenethyl ester (CAPE)
were purchased from Biomol International (Philadelphia, PA,
USA).
Cell culture
The MC3T3-E1 mouse osteoblastic cells (RIKEN Cell
Bank, Tsukuba, Japan) were cultured in plastic dishes containing
α-MEM supplemented with 10% FBS at 37°C in a humidified atmosphere
of 5% CO2 and 95% air. The cells were subcultured every
three days by treating the cells with 0.25% trypsin together with 1
mM EDTA in Ca2+-, Mg2+-free
phosphate-buffered saline (PBS). For immunofluorescence, MC3T3-E1
cells were grown on sterile 18-mm round glass coverslips and
cultured for the desired periods.
RNA preparation and quantitative
polymerase chain reaction (qPCR) assay
Following the appropriate treatment, total cellular
RNA was isolated from MC3T3-E1 cells using RNAiso Plus (Takara Bio,
Inc., Shiga, Japan), followed by phenol extraction and ethanol
precipitation. The purified RNA was further incubated with DNase I
(Sigma-Aldrich) to digest the contaminating DNA. cDNA was
synthesized using ReverTra Ace® qPCR RT Master mix
(Toyobo, Tokyo, Japan). qPCR was conducted using SYBR®
Select Master mix (Applied Biosystems, New York, NY, USA).
Amplified reactions were quantified on an ABI 7500 real-time PCR
system (Applied Biosystems). Relative gene quantities were obtained
using the comparative Ct method following normalization to the
appropriate control genes (β-actin). qPCR was performed on the cDNA
with the following primers: Forward: 5′-CTTTGGGAAACGAGAATTTGGAGA-3′
and reverse: 5′-GCAATCCTG TAGTTGATGGGGAAG-3′ for mouse IL-34; and
forward: 5′-CAATAGTGATGACCTGGCCGT-3′ and reverse:
5′-AGAGGGAAATCGTGCGTGAC-3′ for mouse β-actin.
Immunocytochemistry
Cells were grown on sterile 18-mm round glass cover
slips placed in 60-mm plastic dishes and treated with 0, 1 and 10
ng/ml TNF-α for 0, 15, 30 and 60 min. Additionally, cells were
pretreated with 100 μM CAPE for 1 h and treated with 10 ng/ml TNF-α
for 15 min. The coverslips were washed three times with PBS and
fixed with 3.7% formaldehyde for 10 min at ambient temperature,
followed by methanol-permeabilization for an additional 20 min at
−20°C. Non-specific binding sites were blocked with 4% bovine serum
albumin (BSA) in PBS for 20 min at ambient temperature. The
coverslips were incubated with anti-p65 NF-κB antibody diluted
1:500 for 45 min at ambient temperature. The cells were then
incubated for 30 min with Alexa 488-conjugated goat anti-rabbit IgG
(Invitrogen Life Technologies) diluted 1:500 in 4% BSA in PBS,
followed by incubation for 15 min with 10 μg Hoechst 33342 diluted
1:500 for nucleus staining at ambient temperature. The coverslips
were washed with PBS and mounted with fluorescent mounting medium
(DakoCytomation, Carpinteria, CA, USA). The samples were examined
under an Olympus BX50 microscope (Olympus, Tokyo, Japan) equipped
with epifluorescence illumination. Photomicrographs were recorded
on a computer (DP70-WPCXP; Olympus).
Fractionation of the nucleus and cytosol,
and western blot analysis
Following treatment with 10 ng/ml TNF-α for 0 and 15
min, cells cultured in 90-mm plastic dishes were washed twice with
PBS, collected and resuspended in hypotonic buffer (20 mM HEPES, pH
7.2; 10 mM KCl, 1 mM MgCl2, 1 mM DTT and 0.5 mM EDTA).
Cells were allowed to swell for 10 min in ice prior to lysis by
addition of 0.1% NP-40 and 100 mM potassium acetate. Following 5
min incubation on ice, the cytosolic fraction was recovered in the
supernatant after centrifugation (10,000 × g for 5 min at 4°C). The
pelleted nuclei were resuspended in lysate buffer containing 1 mM
DTT, 1 mM PMSF, 1 mg/ml leupeptin, 2 mg/ml aprotinin and 5 mM EGTA
in PBS, and following centrifugation (20,000 × g for 10 min at
4°C). The nuclear fraction was recovered in the supernatant. The
protein concentration of each sample was evaluated using the
protein assay reagent (Bio-Rad, Hercules, CA, USA). A total of 12
μg of each sample and prestained molecular weight markers were
separated by SDS-PAGE and transferred to polyvinylidene fluoride
membranes (Immobilon-P; Millipore, Bedford, MA, USA). The membranes
were blocked in 5% skimmed milk in PBS-Tween-20 for 2 h. The
membranes were incubated in PBS-Tween-20 containing anti-p65 NF-κB
antibody (diluted at 1:1,000) overnight at 4°C followed by
incubation for 2 h at ambient temperature with an anti-rabbit
horseradish peroxidase-linked secondary antibody (diluted at
1:5,000; Cell Signaling Technology, Inc., Danvers, MA, USA). The
reaction was visualized with an enhanced chemiluminscence detection
kit (GE Healthcare, Chalfont, UK) according to the manufacturer’s
instructions.
Dual-luciferase reporter assay
The luciferase plasmid pNF-κB-Luc was obtained from
Stratagene (La Jolla, CA, USA). MC3T3-E1 cells were seeded into
35-mm plates at a density of 2.0×105 cells/well.
Following 24 h, the cells were co-transfected with 1 μg of
pNF-κB-Luc and 0.05 μg pRL-TK Renilla luciferase vector
(Promega Corporation, Madison, WI, USA) with the aid of the
Lipofectamine reagent. Following 24 h the cells were treated with
or without TNF-α for the indicated duration. The cells were
harvested and treated with passive lysis buffer according to the
dual-luciferase assay manufacturer’s instructions (Promega
Corporation). The signals of firefly luciferase activity were
normalized with respect to pRL-TK Renilla luciferase signals
for individual analysis to eliminate the variations of transfection
efficiencies. Data were analyzed by analysis of variance (ANOVA)
and Bonferroni/Dunn’s test was utilized to estimate the
significance between the means.
Statistical analysis
Each series of experiments were repeated at least
three times and the data are expressed as mean values ± standard
error of mean. Statistical analysis was performed by ANOVA.
P<0.05 was considered to indicate a statistically significant
difference.
Results
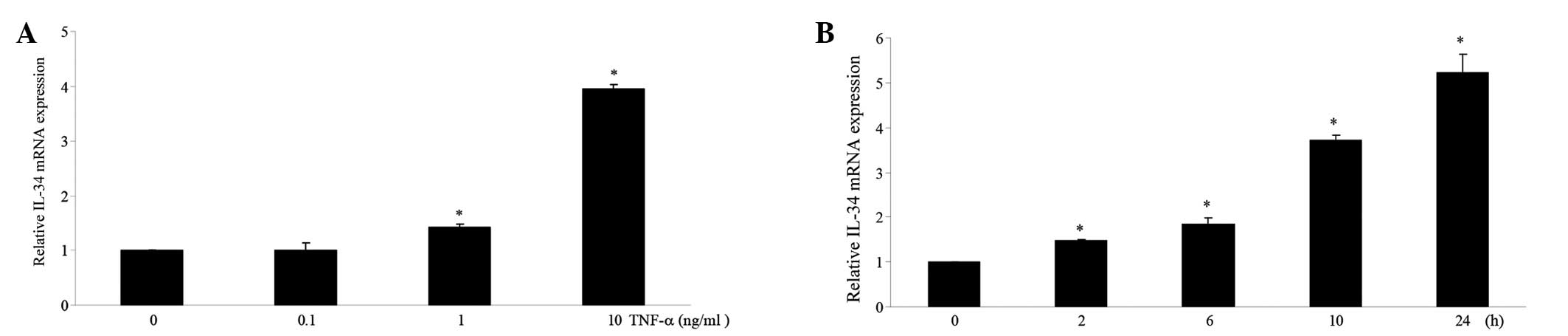
TNF-α increases IL-34 mRNA expression in
a dose- and time-dependent manner in MC3T3-E1 cells
To examine the effect of TNF-α on IL-34 mRNA
expression in mouse osteoblastic cells, MC3T3-E1 cells were treated
with different doses of TNF-α. RNA was collected from the treated
cells and subjected to qPCR using specific primer pairs as
indicated in the Materials and methods. Treatment with TNF-α
increased IL-34 mRNA expression in a dose-dependent manner
(Fig. 1A). The expression of IL-34
mRNA was also increased in a time-dependent manner by TNF-α
treatment (Fig. 1B).
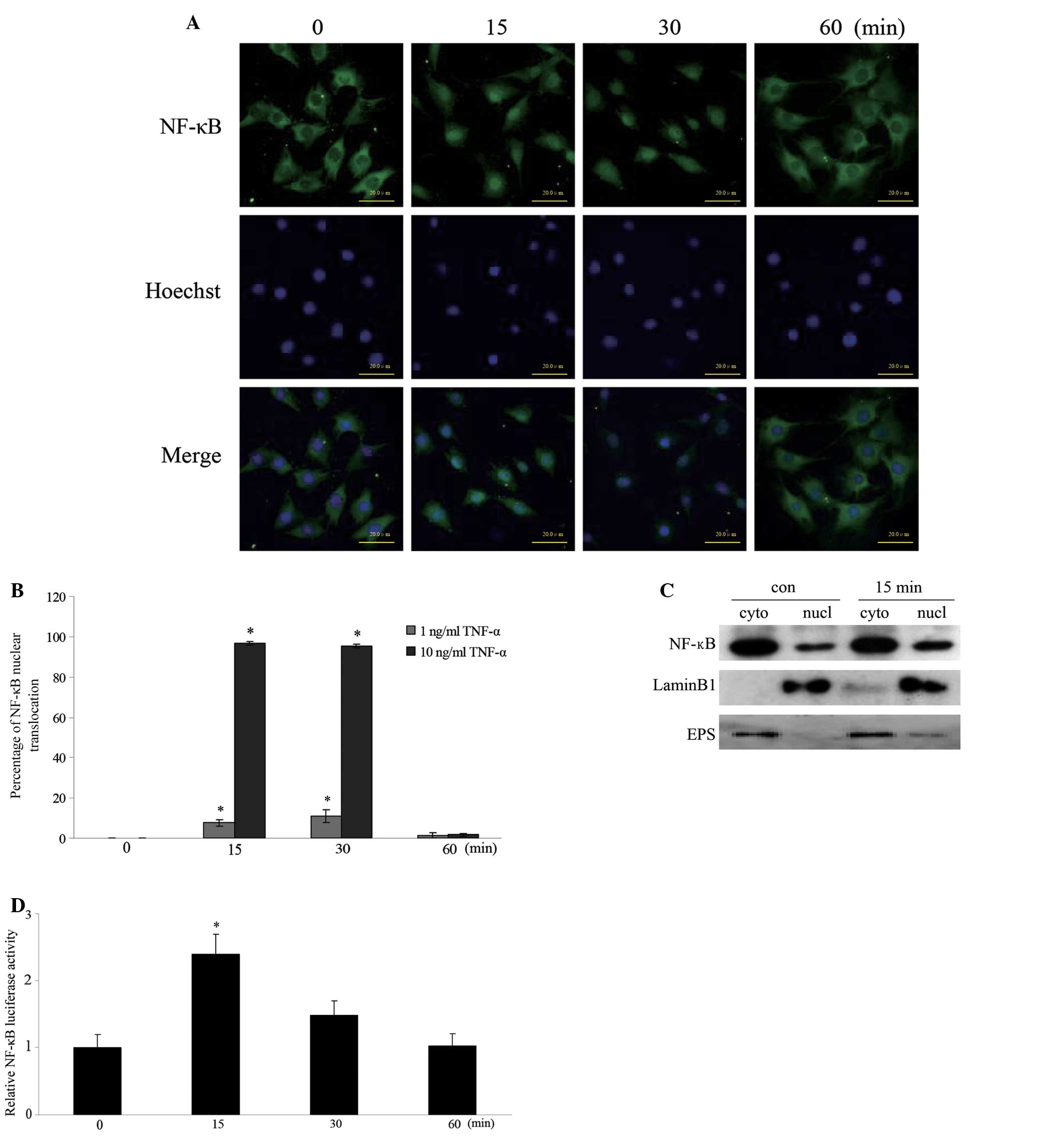
TNF-α induces translocation and
activation of NF-κB in MC3T3-E1 cells
To examine whether TNF-α treatment altered the
subcellular localization of NF-κB, MC3T3-E1 cells were incubated
with 10 ng/ml TNF-α for 0, 15, 30 and 60 min. Fig. 2A demonstrates that NF-κB was mainly
localized in the cytoplasm in the untreated cells. Rapid
translocation of NF-κB into the nucleus was observed in the cells
treated with TNF-α for 15 and 30 min. Fig. 2B reveals the percentages of nuclear
translocation of NF-κB in the cells treated with 1 and 10 ng/ml
TNF-α. The percentages of nuclear translocation of NF-κB treated
with 1 ng/ml TNF-α for 15 and 30 min were 7.6±1.59 and 11.3±3.16%,
respectively. However, the percentages of nuclear translocation of
NF-κB treated with 10 ng/ml TNF-α for 15 and 30 min were 96.6±0.88
and 95.4±0.90%, respectively. To further determine whether TNF-α
induced NF-κB translocation, cell fractionation was performed using
the cells treated with 10 ng/ml TNF-α for 15 min. Fig. 2C demonstrates that the intensity of
the band corresponding to NF-κB in the nuclear fraction was
increased following TNF-α treatment for 15 min compared with that
of the unstimulated cells. The purity of nuclear and cytosolic
fractions was confirmed using an antibody against Lamin B1 (middle)
and anti-Eps15 antibody (bottom), respectively. To further examine
whether TNF-α regulates NF-κB transcriptional activity, the
luciferase reporter assay was performed. TNF-α treatment for 15 min
increased the luciferase activity >2-fold compared with that of
the control cells (Fig. 2D). These
results indicate that TNF-α stimulates NF-κB nuclear translocation
and transcriptional activity.
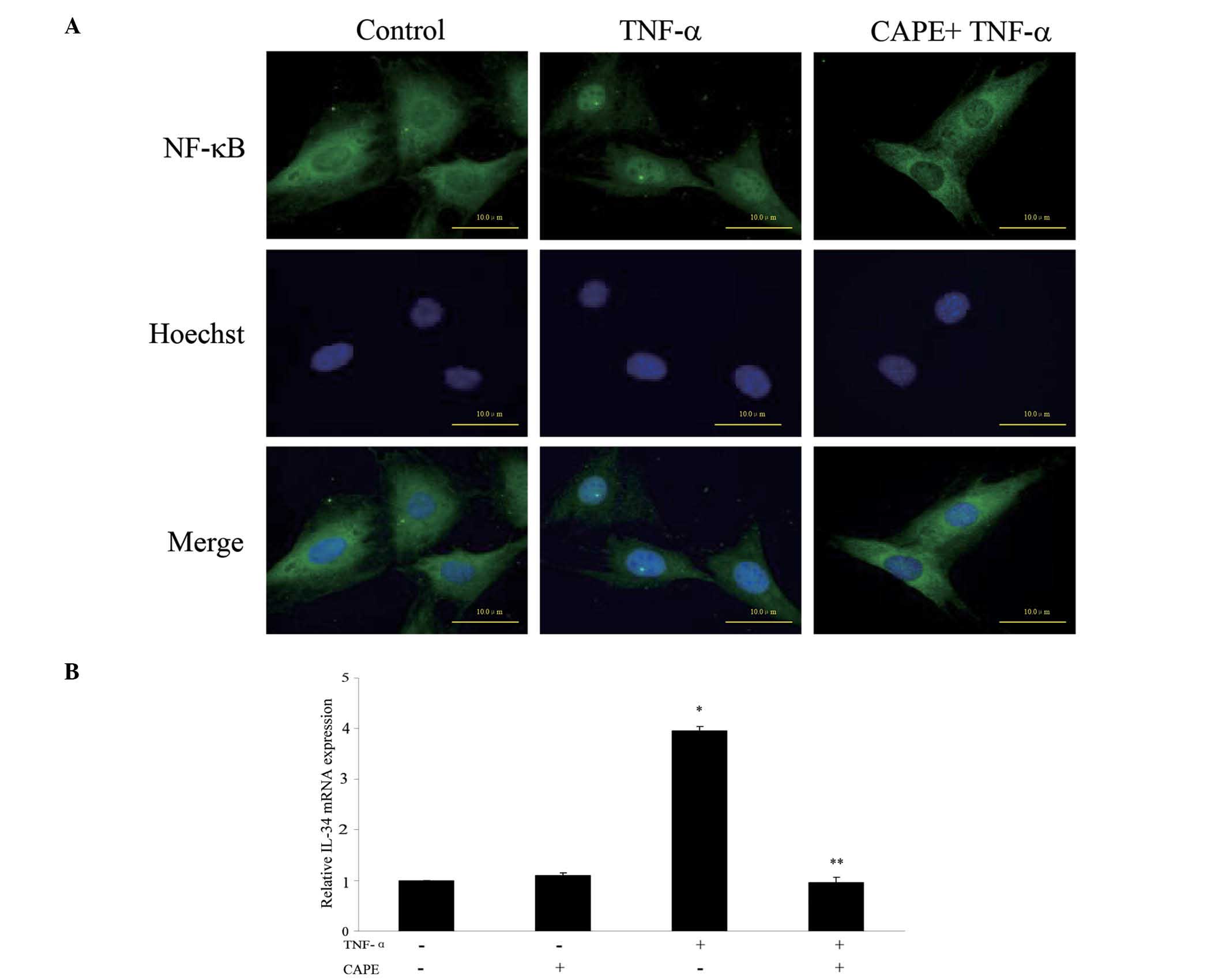
TNF-α increases IL-34 expression via the
NF-κB-dependent pathway in MC3T3-E1 cells
To examine whether NF-κB is involved in the IL-34
expression induced by TNF-α, the cells were treated with TNF-α
following pretreatment with CAPE, an inhibitor of NF-κB. The
nuclear translocation of NF-κB induced by TNF-α was inhibited by
CAPE (Fig. 3A). Pretreatment with
100 μM CAPE for 1 h significantly inhibited TNF-α-induced IL-34
expression (Fig. 3B). However,
treatment with CAPE alone did not change the TNF-α-induced IL-34
expression (Fig. 3B).
Discussion
In the present study, the effect of TNF-α on IL-34
expression and NF-κB activation in osteoblasts was examined. It was
demonstrated that TNF-α induced IL-34 expression in MC3T3-E1 cells
and NF-κB was involved in the TNF-α-induced IL-34 expression in
these cells.
Experimental and clinical studies have demonstrated
that TNF-α is an important factor for bone resorption resulting
from periodontal and periapical diseases (2–6,18).
Elevated TNF-α levels in infected root canals, gingival crevicular
fluid and saliva of patients with aggressive periodontitis has been
reported to stimulate osteoclast generation through the induction
of M-CSF (8,19). Although IL-34 was demonstrated to
have a similar function to M-CSF, the correlation between TNF-α and
the expression of IL-34 in osteoblasts has not been clarified. In
the present study, it was demonstrated that TNF-α treatment induced
IL-34 expression in MC3T3-E1 cells in a dose- and time-dependent
manner. IL-34 is a novel cytokine, which binds to the M-CSF
receptor and possesses similar characteristics to M-CSF in
promoting monocyte viability and osteoclast generation (11,12).
These observations suggest that IL-34 produced from osteoblasts
stimulates osteoclast generation, which leads to bone resorption in
the inflammatory regions.
Following this, the present study aimed to clarify
the mechanism of IL-34 expression induced by TNF-α treatment in
osteoblasts. It was demonstrated that TNF-α treatment rapidly
induced NF-κB translocation from the cytoplasm into the nucleus in
MC3T3-E1 cells, as determined by immunostaining and cell
fractionation assays. Consistent with these results, the luciferase
assay revealed that NF-κB transcriptional activity was
significantly increased in MC3T3-E1 cells treated with TNF-α. It
was reported that TNF-α induced M-CSF expression in primary
osteoblasts and osteoblastic cells through the activation of NF-κB
(20,21). Therefore, it was hypothesized that
NF-κB is involved in the IL-34 expression in MC3T3-E1 cells induced
by TNF-α. To verify this hypothesis, MC3T3-E1 cells were pretreated
with NF-κB specific inhibitor CAPE, followed by TNF-α treatment for
15 min. Pretreatment of 100 μM CAPE markedly inhibited
TNF-α-induced IL-34 expression in MC3T3-E1 cells, indicating that
NF-κB is involved in the expression of IL-34 in these cells.
Previously, it was reported that TNF-α-induced IL-34 expression in
synovial fibroblasts was mediated through the activation of NF-κB
(22,23). NF-κB mediates the expression of
numerous inflammatory genes, including IL-6 and ICAM-1, in rat and
human osteoblast-like cells, such as UMR106 and MG63 cells
(16,17,24,25).
By contrast, it was reported that lipopolysaccharide and
interferon-γ only moderately increased IL-34 mRNA levels, although
these factors were well-known stimuli for eliciting a variety of
inflammatory responses (26). The
present results, coupled with the previous evidence, suggests that
NF-κB is an important mediator for regulating the expression of
inflammatory cytokines induced by TNF-α in osteoblastic cells.
In conclusion, the data demonstrated in the present
study provides evidence that TNF-α induces IL-34 expression via
NF-κB in MC3T3-E1 cells. Further investigation is required to
define the pathological implications of IL-34 produced from
osteoblasts in local inflammatory regions, including in periodontal
and periapical diseases. Studies designed to examine the potential
of the NF-κB pathway as a therapeutic target for these inflammatory
conditions in vivo are also warranted.
References
|
1
|
Braun T and Schett G: Pathways for bone
loss in inflammatory disease. Curr Osteoporos Rep. 10:101–108.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gümüş P, Nizam N, Lappin DF and Buduneli
N: Saliva and serum levels of B-cell activating factors and tumor
necrosis factor-α in patients with periodontitis. J Periodontol.
85:270–280. 2014.
|
|
3
|
Jiang ZL, Cui YQ, Gao R, Li Y, Fu ZC,
Zhang B and Guan CC: Study of TNF-α, IL-1β and LPS levels in the
gingival crevicular fluid of a rat model of diabetes mellitus and
periodontitis. Dis Markers. 34:295–304. 2013.
|
|
4
|
Yue Y, Liu Q, Xu C, et al: Comparative
evaluation of cytokines in gingival crevicular fluid and saliva of
patients with aggressive periodontitis. Int J Biol Markers.
28:108–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martinho FC, Chiesa WM, Leite FR, Cirelli
JA and Gomes BP: Correlation between clinical/radiographic features
and inflammatory cytokine networks produced by macrophages
stimulated with endodontic content. J Endod. 38:740–745. 2012.
View Article : Google Scholar
|
|
6
|
Oliveira LD, Carvalho CA, Carvalho AS, de
Alves JS, Valera MC and Jorge AO: Efficacy of endodontic treatment
for endotoxin reduction in primarily infected root canals and
evaluation of cytotoxic effects. J Endod. 38:1053–1057. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chitu V and Stanley ER: Colony-stimulating
factor-1 in immunity and inflammation. Curr Opin Immunol. 18:39–48.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Felix R, Fleisch H and Elford PR:
Bone-resorbing cytokines enhance release of macrophage
colony-stimulating activity by the osteoblastic cell MC3T3-E1.
Calcif Tissue Int. 44:356–360. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tanaka H, Tanabe N, Shoji M, et al:
Nicotine and lipopolysaccharide stimulate the formation of
osteoclast-like cells by increasing macrophage colony-stimulating
factor and prostaglandin E2 production by osteoblasts. Life Sci.
78:1733–1740. 2006. View Article : Google Scholar
|
|
10
|
Katono T, Kawato T, Tanabe N, et al:
Nicotine treatment induces expression of matrix metalloproteinases
in human osteoblastic Saos-2 cells. Acta Biochim Biophys Sin
(Shanghai). 38:874–882. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin H, Lee E, Hestir K, et al: Discovery
of a cytokine and its receptor by functional screening of the
extracellular proteome. Science. 320:807–811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Z, Buki K, Vääräniemi J, Gu G and
Väänänen HK: The critical role of IL-34 in osteoclastogenesis. PLoS
One. 6:e186892011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boyce BF, Yao Z and Xing L: Functions of
nuclear factor kappaB in bone. Ann NY Acad Sci. 1192:367–375. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Novack DV: Role of NF-κB in the skeleton.
Cell Res. 21:169–182. 2011.
|
|
15
|
Wang T, Zhang X and Li JJ: The role of
NF-kappaB in the regulation of cell stress responses. Int
Immunopharmacol. 2:1509–1520. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kobayashi K, Kambe F, Kurokouchi K, et al:
TNF-alpha-dependent activation of NF-kappa B in human osteoblastic
HOS-TE85 cells is repressed in vector-averaged gravity using
clinostat rotation. Biochem Biophys Res Commun. 279:258–264. 2000.
View Article : Google Scholar
|
|
17
|
Kurokouchi K, Kambe F, Yasukawa K, Izumi
R, Ishiguro N, Iwata H and Seo H: TNF-alpha increases expression of
IL-6 and ICAM-1 genes through activation of NF-kappaB in
osteoblast-like ROS17/2.8 cells. J Bone Miner Res. 13:1290–1299.
1998. View Article : Google Scholar
|
|
18
|
Boyce BF, Li P, Yao Z, et al: TNF-alpha
and pathologic bone resorption. Keio J Med. 54:127–131. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sato K, Kasono K, Fujii Y, Kawakami M,
Tsushima T and Shizume K: Tumor necrosis factor type alpha
(cachectin) stimulates mouse osteoblast-like cells (MC3T3-E1) to
produce macrophage colony stimulating activity and prostaglandin
E2. Biochem Biophys Res Commun. 145:323–329. 1987. View Article : Google Scholar
|
|
20
|
Yao GQ, Sun BH, Insogna KL and Weir EC:
Nuclear factor-kappaB p50 is required for tumor necrosis
factor-alpha-induced colony-stimulating factor-1 gene expression in
osteoblasts. Endocrinology. 141:2914–2922. 2000.PubMed/NCBI
|
|
21
|
Eda H, Shimada H, Beidler DR and Monahan
JB: Proinflammatory cytokines, IL-1β and TNF-α, induce expression
of interleukin-34 mRNA via JNK- and p44/42 MAPK-NF-κB pathway but
not p38 pathway in osteoblasts. Rheumatol Int. 31:1525–1530.
2011.
|
|
22
|
Chemel M, Le Goff B, Brion R, et al:
Interleukin 34 expression is associated with synovitis severity in
rheumatoid arthritis patients. Ann Rheum Dis. 71:150–154. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hwang SJ, Choi B, Kang SS, et al:
Interleukin-34 produced by human fibroblast-like synovial cells in
rheumatoid arthritis supports osteoclastogenesis. Arthritis Res
Ther. 14:R142012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kurokouchi K, Jacobs CR and Donahue HJ:
Oscillating fluid flow inhibits TNF-alpha-induced NF-kappa B
activation via an Ikappa B kinase pathway in osteoblast-like UMR106
cells. J Biol Chem. 276:13499–13504. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kurokouchi K, Kambe F, Kikumori T, et al:
Effects of glucocorticoids on tumor necrosis factor alpha-dependent
activation of nuclear factor kappaB and expression of the
intercellular adhesion molecule 1 gene in osteoblast-like ROS17/2.8
cells. J Bone Miner Res. 15:1707–1715. 2000. View Article : Google Scholar
|
|
26
|
Wei S, Nandi S, Chitu V, et al: Functional
overlap but differential expression of CSF-1 and IL-34 in their
CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol.
88:495–505. 2010. View Article : Google Scholar : PubMed/NCBI
|