Introduction
Malignant melanoma (MM) is an aggressive,
therapy-resistant malignancy of the melanocytes. The incidence of
MM has been steadily increasing worldwide, resulting in an
increasing public health problem (1). More than 95% of the MM tumors occur
in the skin. MM is ranked as the third most common type of
cutaneous malignant tumor, with a high malignance and high
potential of hematogenous and lymphatica metastasis. During the
progression and therapy of MM, melanoma cell lines demonstrate
increased resistance to death receptor-mediated apoptosis and
drug-induced apoptosis compared with their normal counterparts
(2).
Apoptosis induced by antineoplastic drugs was not
affected by the absence of Fas, indicating that cell death was in a
Fas-independent manner. The insensitive manner pointed to
FLIP-induced apoptosis (3),
indicating the existence of another pathway responsible for
preventing apoptosis.
Cellular caspase-8 (FLICE)-like inhibitory protein
(c-FLIP) was originally identified as an inhibitor of
death-receptor signaling through competition with caspase-8 for
recruitment to FAS-associated protein with death domain (FADD).
c-FLIPL, the long isoform of c-FLIP is specifically
processed by caspase 8 into N-terminal c-FLIPp43 and
C-terminal c-FLIPp12. In accordance with others studies,
we previously identified that compared with the pigmented nevi
lesions, the expression of c-FLIP increased and was significantly
associated with the histological type and Clark’s level in MM
tissue samples (4–6). It was also identified that the
downregulated expression of c-FLIP increases apoptosis in MM cell
lines. However, the molecular mechanisms of c-FLIP in melanoma
pathogenesis remain unclear.
Dohrman and Kataoka et al (7,8)
reported that c-FLIPL may be cleaved into
c-FLIPp43, promoting the activation of nuclear factor
(NF)-κB in CD8+ T cells, human embryonic kidney (HEK)
293 cells and 293 T cells. NF-κB is a pro-inflammatory
transcription factor and is activated by various inflammatory
agents, carcinogens, tumor promoters and the tumor
microenvironment. NF-κB protein, and the proteins regulated by
NF-κB are associated with cellular transformation, proliferation,
apoptosis suppression, invasion, angiogenesis and metastasis during
the genesis of tumors (9). NF-κB
is expressed and is constitutively active in the majority of tumor
cell lines, including melanoma cells (10). The high level of non-canonical
NF-κB activation could be decreased by NIK depletion and thus
resulted in increased apoptosis (11).
It remains unclear whether c-FLIPp43 is
involved in the pathogenesis of melanoma through NF-κB activation.
The aim of the present study was to evaluate whether the regulation
of c-FLIPp43 induces the NF-κB signaling pathway in
melanoma cell lines and subsequently promotes the progression of
melanoma. The present study investigated the expression profile of
c-FLIPp43 and NF-κB in melanoma cell lines by
immunoblotting. In addition, a eukaryotic expression vector for
c-FLIPp43 and a monoclonal A375 cell line with the
stable expression of c-FLIPp43 were successfully
constructed. Immunofluorescent staining was also utilized to
investigate the nuclear translocation of NF-κBp65 with
the stable expression of c-FLIPp43 and a dual-luciferase
reporter assay system was used to investigate the NF-κB
transactivation induced by different c-FLIPp43 vector
concentrations in the A375 cell line.
Materials and methods
Melanoma cell lines, expression vectors,
antibodies and reagents
The A375 and SK-Mel-1 human malignant melanoma cell
lines (Institute of Cell Biology, Shanghai Institute for Biological
Science, Chinese Academy of Science, Shanghai, China) were cultured
in plastic flasks in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco-BRL, Manheim, Germany), containing 10% heat-inactivated fetal
calf serum (Hyclone, Logan, UT, USA), 15 mm HEPES, 2 mml-glutamine,
and 100 U/ml penicillin and streptomycin at 37°C.
pCMV-Tag2B-cFLIPp43 and pCMV-Tag2B were constructed by
the Laboratory of Molecular Biology in Hubei University (Hubei,
China). pGL3-3xκB-Luc and pRL-TK were provided by Professor Lixin
Ma’s experimental group of Hubei University. Antibodies against
c-FLIP, NF-κBp65 and β-actin were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The NF-κB
Activation, Nuclear Translocation Assay kit and Nuclear and
Cytoplasmic Protein Extraction kit were obtained from Biyuntian
Company (Beijing, China). Lipofectamine™ 2000 was purchased from
Invitrogen Life Technologies (San Diego, CA, USA).
Cell transfection and selection of stable
G418 resistant clones
pCMV-Tag2B-cFLIPp43 was transfected into
A375 cells with Lipofectamine 2000, and 48 h following
transfection, cells were treated daily with 500 mg/ml G418
(Invitrogen Life Technologies) as a selective marker for two weeks.
The surviving cells were dispersed at a density of one cell/well in
24-well multiwell plates and several stably transfected cell clones
were obtained in 3–4 weeks. The stable clones were screened for
pCMV-Tag2B-cFLIPp43 expression by western blotting.
Western blotting
A total of 1×106 cells were incubated for
30 min at 4°C in 100 μl radioimmunoprecipitation assay buffer (cell
lysis buffer for western blotting and IP). A total of 30 μg of
protein per slot was separated by discontinuous sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, with the gel containing
12% acrylamide. Following blotting onto a polyvinylidene difluoride
membrane (Millipore, Billerica, MA, USA), 1 h incubation with
non-fat milk was performed at 37°C using a shaker (WD-9405B;
Beijing Liuyi Instrument Factory, Beijing, China) to inhibit the
unspecific binding of antibodies. The blots were incubated
overnight in a 4°C icebox with the primary antibody. Subsequently,
the blots were washed three times each for 10 min with
Tris-buffered saline containing 0.1% Tween-20 and a secondary
antibody conjugated to horseradish peroxidase (dilution factor,
1:8000; Dako, Hamburg, Germany) was added. The Enhanced
Chemiluminescence kit (Thermo Fisher Scientific Inc., Rockford, IL,
USA) was used to develop the blots.
Immunofluorescence assay
The cover slips were located in twenty-four-well
culture plates and A375 cells, which were transfected with
pCMV-Tag2B-cFLIPp43, were seeded onto it. The next day,
the culture media were discarded and the cells were washed once
with phosphate-buffered saline (PBS). Paraformaldehyde (4%;
Beyotime Institute of Biotechnology, Nantong, Jiangsu, China) was
added for 10–15 min and then discarded. The cells were washed with
cleaning solution three times for 3–5 min each time. Fetal Bovine
Serum (Gibco-BRL, Carlsbad, CA, USA) was added for 1 h at room
temperature and then discarded. The cells were incubated with
antibodies against NF-κBp65 overnight at 4°C. The cells
were then washed with PBS (Beyotime Institute of Biotechnology)
three times, for 3–5 min each time. The cells were incubated with
anti-rabbit Cy3 (Jackson ImmunoResearch, West Grove, PA, USA) for 1
h at room temperature. The cells were washed twice with cleaning
solution for 5–10 min each time. DAPI staining solution was added
for 5 min at room temperature and then removed. The cells were then
washed with cleaning solution three times for 3–5 min each time.
Anti-fade mounting media (Beyotime Institute of Biotechnology) were
added and the cover slips were located, viewed and photographed
under a laser scanning confocal microscope (Olympus, Center Valley,
PA, USA). NF-κBp65 staining was red and DAPI staining of
the nucleus was blue.
Dual-luciferase reporter
Six-well culture plates were seeded with A375 cells.
At 60–80 degrees of fusion, the cells were transiently transfected
according to the manufacturer’s details of the Effectene
Transfection Reagent kit (Qiagen Inc., Venlo, Netherlands). The
cells were collected 20 h later. The ratio of Firefly luciferase
and Renilla luciferase were analyzed by Turner Bio Systems
Modulus Luminometer (Reporter Microplate Luminometer; Turner
BioSystems Inc., Sunnyvale, CA, USA).
Statistical analysis
Data analysis was performed using the SPSS 11.0
Statistical Software (SPSS, Inc., Chicago, IL, USA). An independent
sample’s t-test was used and P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression of c-FLIP,
c-FLIPp43 and NF-κBp65 in A375 cells
Immunoblotting analysis of the A375 and SK-Mel-1
melanoma cell lines revealed that c-FLIPp43 expression
in the SK-Mel-1 cells was higher than that in the A375 cells
(Fig. 1A). The expression of
c-FLIP was undetectable in A375 cells but high expression was
observed in the SK-Mel-1 cells. The expression of
NF-κBp65 in A375 cells was lower than that in the
SK-Mel-1 cells (Fig. 1B).
Stable expression of c-FLIPp43
and NF-κBp65 in A375 cells stably transfected with
pCMV-Tag2B-cFLIPp43
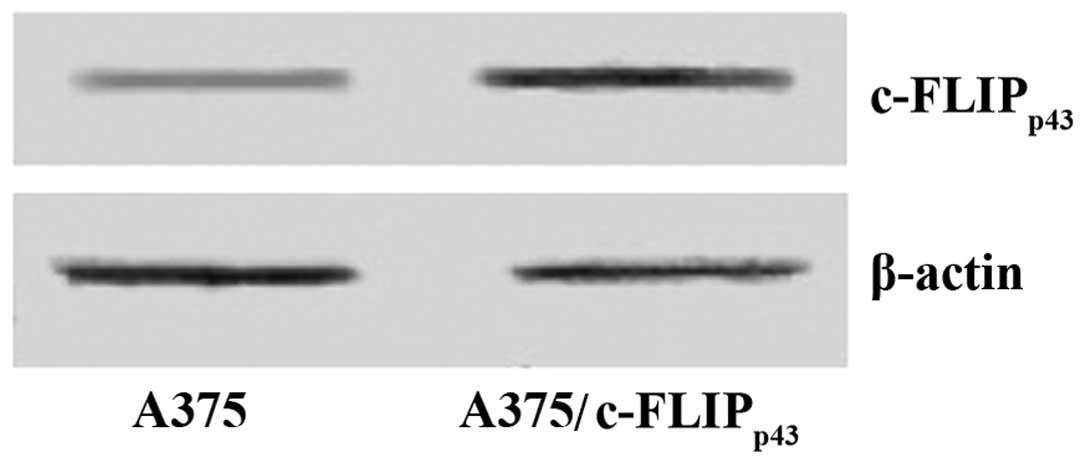
pCMV-Tag2B-cFLIPp43 was transfected into
the A375 cell line and the stable clones were isolated following
G418 selection. To confirm successful transfection of A375 cells
with the c-FLIPp43 construct, immunoblotting analysis
was conducted. The expression level of c-FLIPp43 was
evidently higher in the A375 cells that were stably transfected
with c-FLIPp43, than in the A375 cells, which were not
transfected (Fig. 2). Consistent
with this, the immunoblotting results of A375 cells stably
expressing c-FLIPp43 demonstrated increased expression
of NF-κBp65 (Fig. 3).
These data suggest that overexpression of c-FLIPp43 may
upregulate NF-κBp65 expression at the protein level.
NF-κBp65 nuclear
translocation
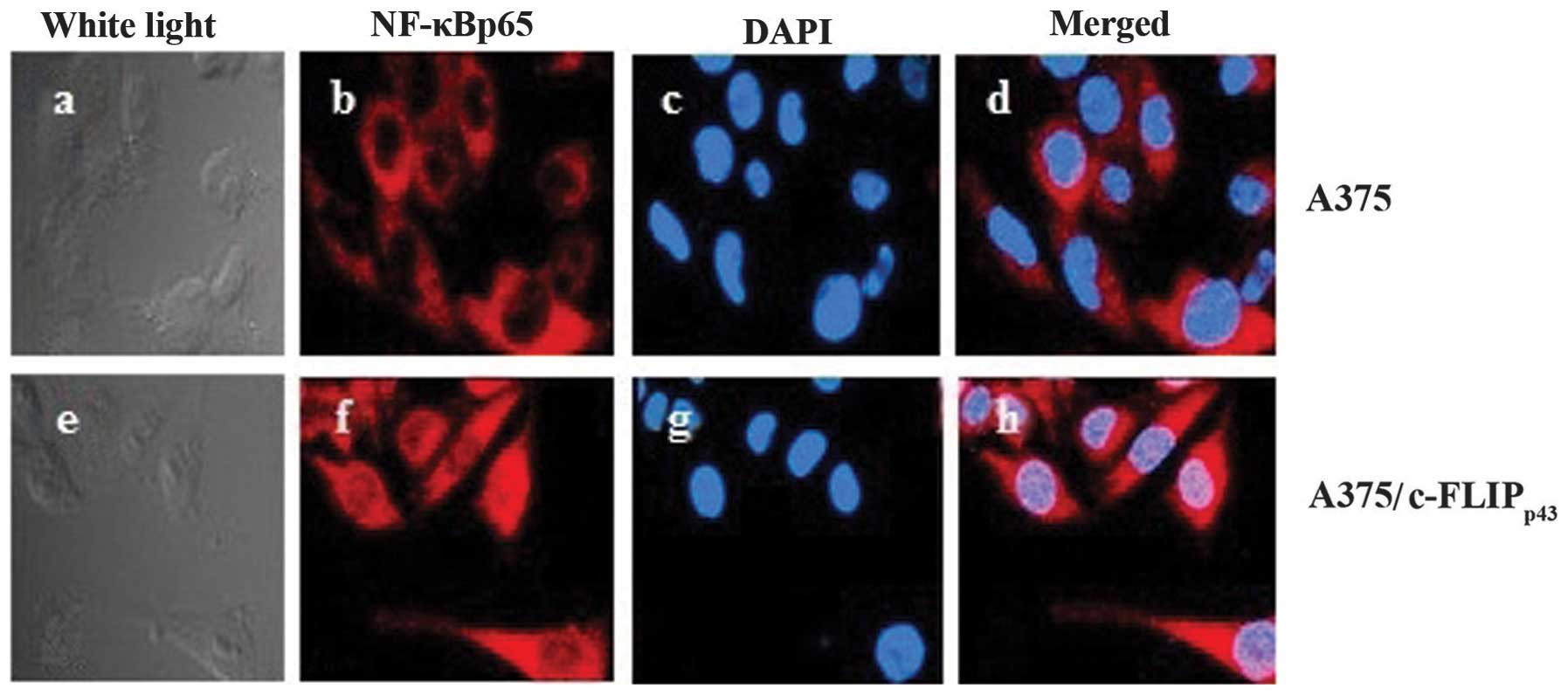
Immunofluorescent staining of NF-κBp65
was performed in the A375 cell line stably expressing
c-FLIPp43. An overlay of merged images
(NF-κBp65 in red and DAPI in blue) demonstrates that
NF-κBp65 was predominantly localized in the nuclei and
plasma in A375 cells transfected with
pCMV-Tag2B-cFLIPp43, while there was weak plasma and
nuclei staining in the untreated cells (Fig. 4). c-FLIPp43 transfection
increased the NF-κBp65 fluorodensitometric value
(P<0.01) compared with the transfection with empty vector
(Table I). Collectively, these
results indicate that NF-κBp65 nuclear translocation was
increased in the A375 cells following transfection with
pCMV-Tag2B-cFLIPp43.
 | Table IFluorodensitometric value of
NF-κBp65 in the nuclei. |
Table I
Fluorodensitometric value of
NF-κBp65 in the nuclei.
| Fluorodensitometric
value | P-value |
|---|
| A375 | 17.34±8.22 | |
| A375/cFLIPp43 | 60.71±18.32 | <0.01 |
NF-κBp65 activation examined
by the dual-luciferase reporter assay
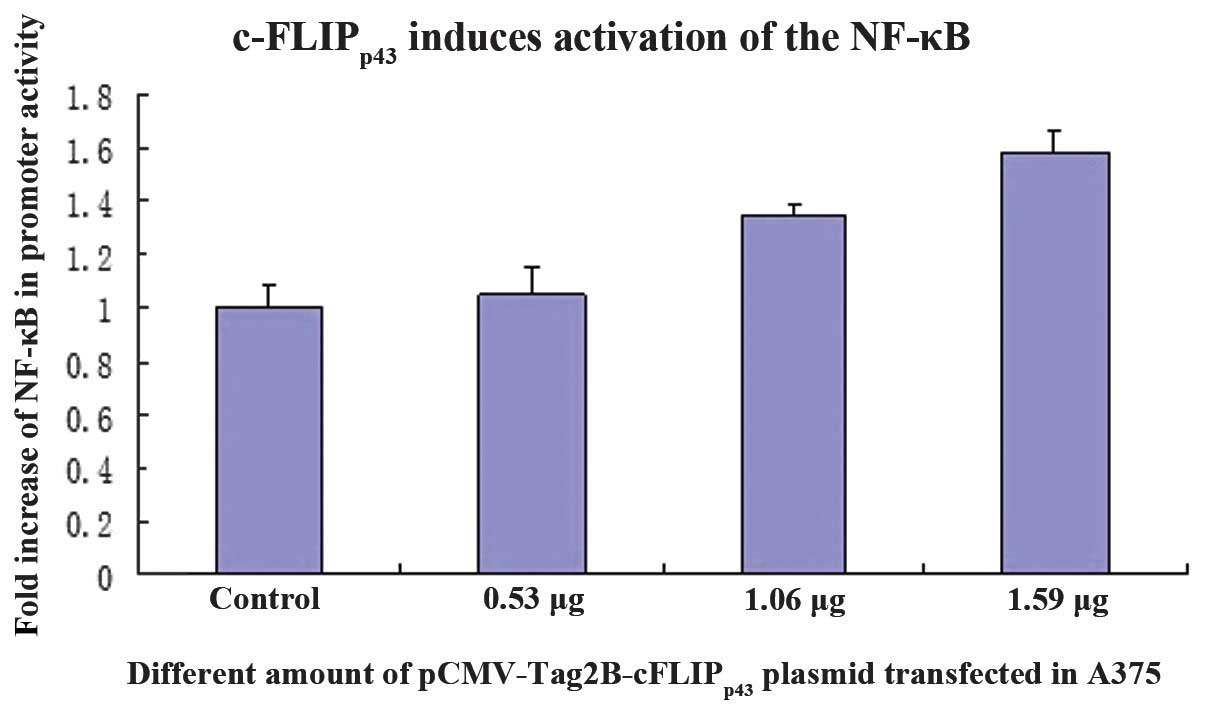
To determine whether c-FLIPp43 was able
to induce NF-κB transactivation, the luciferase activity was
measured using a dual-luciferase reporter assay system. The A375
cells, which were transfected with pCMV-Tag2B-cFLIPp43,
induced a 1.5-fold increase in promoter activity compared with the
control cells (Fig. 5). This
implies that effective c-FLIPp43 expression is required
for NF-κB activation and this increase was in a dose-dependent
manner.
Discussion
c-FLIP has been observed at increased levels in a
number of cell lines or cancerous tissues, including melanoma
(5,6,12),
gastric (13–15), lung (5,16),
colorectal (16), hepatocellular
(17), pancreatic (18), ovarian (19) and prostate carcinoma (20), and B-cell chronic lymphocytic
leukemia (21,22). Elevated levels of c-FLIP have been
demonstrated to correlate with more aggressive tumors and a poor
prognosis (23–28).
Zeise et al (29) found that different melanoma cells
demonstrated sensitivity or resistance towards TNF-related
apoptosis-inducing ligand (TRAIL)-induced apoptosis, which may have
been due to different expression and the pre-apoptotic processing
status of FLIP. The authors also found that A375 cells appeared
sensitive to TRAIL-induced apoptosis and a number others, including
SK-Mel-30, were resistant to TRAIL-induced apoptosis. The present
study demonstrated that the expression of c-FLIPp43 was
low in A375 cells but high in the SK-Mel-1 cells. High c-FLIP
expression has been found to inhibit TRAIL-induced apoptosis by
preventing activation of the caspase cascade (30,31).
Differential sensitivity of cancer cells to TRAIL-induced apoptosis
may be determined by intracellular c-FLIP levels. Though TRAIL is
regarded as a promising anticancer drug due to its high
selectivity, the regulation of c-FLIP may be considered as a
potential strategy in certain TRAIL-resistant cancer cells.
It was noted that the expression of
NF-κBp65 in A375 cells was lower than that in the
SK-Mel-1 cells. It has been reported that besides the crucial role
of c-FLIP in death receptor (DR)-induced apoptosis, the c-FLIP
cleavage product c-FLIPp43 may also regulate DR-induced
NF-κB activity (8,32–34).
Others studies have reported that the regulation of
c-FLIPL in CD95-mediated NF-κB activation may be
concentration-or cell line-dependent and different levels of c-FLIP
expression may promote or inhibit the pathway (35).
To improve the understanding of the role of
c-FLIPp43 in the pathogenesis of melanoma, a eukaryotic
expression vector for c-FLIPp43 with the plasmid
pCMV-Tag2B was constructed and the stable expression of
c-FLIPp43 monoclonal A375 cells was obtained.
It was identified that NF-κB activity was elevated
following transfection with pCMV-Tag2B-cFLIPp43 in the
A375 cells. As the expression of c-FLIPp43 increased,
the NF-κBp65 activity was enhanced. These results
indicated that c-FLIPp43 may promote the activation of
NF-κBp65 in a dose-dependent manner. These observations
were consistent with those of Kataoka et al (3), which found that c-FLIPL
was cleaved into c-FLIPp43 by caspase-8 to promote the
activation of NF-κB. Caspase 8 activity is thus required for
FLIPL-induced NF-κB activation. FLIPL may be
specifically processed by caspase 8 and be cleaved into
FLIPp43 and FLIPp12. In caspase-8 deficient
cells, c-FLIPp43 was not able to provoke NF-κB
activation without complemented procaspase-8 or the intermediate
cleavage products caspase-8p43. FLIPL appears
to have a molecular switch role to regulate cell death and cell
growth by inhibiting the activation of caspase 8, as well as
proliferative signaling pathways, including the NF-κB pathway
(7). Transfection of
c-FLIPp43 promoted NF-κB activation and increased the
expression of NF-κB in A375 cells. This may explain that
overexpression of FLIPp43 maintains the NF-κB activation
and downregulation of c-FLIP induces apoptosis in the A875
malignant melanoma cell line (6).
Melanoma progression is a multistep process. It
involves multiple genetic mutations and multimolecular activation,
and NF-κB has also been considered to be involved (36). It has been reported that NF-κB
activation may inhibit apoptosis in tumor cells and further promote
tumor formation and chemoresistance (37). Huang et al (38) found that the constitutive
expression RelA (p65) a member of the NF-κB family was relevant to
the metastasis of melanoma. These results indicated that
c-FLIPp43 may be one inducer that leads to the
activation of NF-κB and thus promotes melanoma progression.
In conclusion, transfection of the
c-FLIPp43 expression vector may induce the protein
expression of NF-κBp65 and promote the activation of the
NF-κB signaling pathway in the A375 melanoma cell line. Targeting
c-FLIPp43 may be a promising potential strategy for
melanoma treatment, particularly in TRAIL-resistant cell lines.
Furthermore, the combination therapy of c-FLIPp43
regulation with TRAIL-related treatment (39), inhibitors of the NF-κB signaling
pathway or conventional chemotherapy may be an effective treatment
option and requires further investigation to elucidate its clinical
utility.
References
|
1
|
Markovic SN, Erickson LA, Rao RD, et al:
Malignant melanoma in the 21st century, part 1: epidemiology, risk
factors, screening, prevention, and diagnosis. Mayo Clinic Proc.
82:364–380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Igney FH and Krammer PH: Death and
anti-death: tumour resistance to apoptosis. Nature Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kataoka T, Schröter M, Hahne M, et al:
FLIP prevents apoptosis induced by death receptors but not by
perforin/granzyme B, chemotherapeutic drugs, and gamma irradiation.
J Immunol. 161:3936–3942. 1998.
|
|
4
|
Irmler M, Thome M, Hahne M, et al:
Inhibition of death receptor signals by cellular FLIP. Nature.
388:190–195. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bullani RR, Huard B, Viard-Leveugle I, et
al: Selective expression of FLIP in malignant melanocytic skin
lesions. J Invest Dermatol. 117:360–364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tian F, Lu JJ, Wang L, et al: Expression
of c-FLIP in malignant melanoma, and its relationship with the
clinicopathological features of the disease. Clin Exp Dermatol.
37:259–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kataoka T and Tschopp J: N-terminal
fragment of c-FLIP(L) processed by caspase 8 specifically interacts
with TRAF2 and induces activation of the NF-kappaB signaling
pathway. Mol Cell Biol. 24:2627–2636. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dohrman A, Kataoka T, Cuenin S, et al:
Cellular FLIP (long form) regulates CD8+ T cell
activation through caspase-8-dependent NF-kappa B activation. J
Immunol. 174:5270–5278. 2005.PubMed/NCBI
|
|
9
|
Aggarwal BB: Nuclear factor-kappa B: the
enemy within. Cancer Cell. 6:203–208. 2004.
|
|
10
|
McNulty SE, del Rosario R, Cen D, Meyskens
FL Jr and Yang S: Comparative expression of NFkappaB proteins in
melanocytes of normal skin vs. benign intradermal naevus and human
metastatic melanoma biopsies. Pigment Cell Res. 17:173–180. 2004.
View Article : Google Scholar
|
|
11
|
Thu YM, Su Y, Yang J, et al: NF-kappaB
inducing kinase (NIK) modulates melanoma tumorigenesis by
regulating expression of pro-survival factors through the β-catenin
pathway. Oncogene. 31:2580–2592. 2012.PubMed/NCBI
|
|
12
|
Griffith TS, Chin WA, Jackson GC, Lynch DH
and Kubin MZ: Intracellular regulation of TRAIL-induced apoptosis
in human melanoma cells. J Immunol. 161:2833–2840. 1998.PubMed/NCBI
|
|
13
|
Lee SH, Kim HS, Kim SY, et al: Increased
expression of FLIP, an inhibitor of Fas-mediated apoptosis, in
stomach cancer. APMIS. 111:309–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nam SY, Jung GA, Hur GC, et al:
Upregulation of FLIP(S) by Akt, a possible inhibition mechanism of
TRAIL-induced apoptosis in human gastric cancers. Cancer Science.
94:1066–1073. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou XD, Yu JP, Liu J, et al:
Overexpression of cellular FLICE-inhibitory protein (FLIP) in
gastric adenocarcinoma. Clin Sci (Lond). 106:397–405. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wilson NS, Dixit V and Ashkenazi A: Death
receptor signal transducers: nodes of coordination in immune
signaling networks. Nat Immunol. 10:348–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okano H, Shiraki K, Inoue H, et al:
Cellular FLICE/caspase-8-inhibitory protein as a principal
regulator of cell death and survival in human hepatocellular
carcinoma. Lab Invest. 83:1033–1043. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elnemr A, Ohta T, Yachie A, et al: Human
pancreatic cancer cells disable function of Fas receptors at
several levels in Fas signal transduction pathway. Int J Oncol.
18:311–316. 2001.
|
|
19
|
Xiao CW, Yan X, Li Y, Reddy SA and Tsang
BK: Resistance of human ovarian cancer cells to tumor necrosis
factor alpha is a consequence of nuclear factor kappaB-mediated
induction of Fas-associated death domain-like
interleukin-1beta-converting enzyme-like inhibitory protein.
Endocrinology. 144:623–630. 2003. View Article : Google Scholar
|
|
20
|
Zhang X, Jin TG, Yang H, et al: Persistent
c-FLIP(L) expression is necessary and sufficient to maintain
resistance to tumor necrosis factor-related apoptosis-inducing
ligand-mediated apoptosis in prostate cancer. Cancer Res.
64:7086–7091. 2004. View Article : Google Scholar
|
|
21
|
Olsson A, Diaz T, Aguilar-Santelises M, et
al: Sensitization to TRAIL-induced apoptosis and modulation of
FLICE-inhibitory protein in B chronic lymphocytic leukemia by
actinomycin D. Leukemia. 15:1868–1877. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
MacFarlane M, Harper N, Snowden RT, et al:
Mechanisms of resistance to TRAIL-induced apoptosis in primary B
cell chronic lymphocytic leukaemia. Oncogene. 21:6809–6818. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Korkolopoulou P, Goudopoulou A, Voutsinas
G, et al: c-FLIP expression in bladder urothelial carcinomas: its
role in resistance to Fas-mediated apoptosis and clinicopathologic
correlations. Urology. 63:1198–1204. 2004. View Article : Google Scholar
|
|
24
|
Djerbi M, Screpanti V, Catrina AI, et al:
The inhibitor of death receptor signaling, FLICE-inhibitory protein
defines a new class of tumor progression factors. J Exp Med.
190:1025–1032. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ullenhag GJ, Mukherjee A, Watson NF, et
al: Overexpression of FLIPL is an independent marker of poor
prognosis in colorectal cancer patients. Clin Cancer Res.
13:5070–5075. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang W, Wang S, Song X, et al: The
relationship between c-FLIP expression and human papillomavirus E2
gene disruption in cervical carcinogenesis. Gynecol Oncol.
105:571–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Valnet-Rabier MB, Challier B, Thiebault S,
et al: c-Flip protein expression in Burkitt’s lymphomas is
associated with a poor clinical outcome. Br J Haematol.
128:767–773. 2005.
|
|
28
|
Valente G, Manfroi F, Peracchio C, et al:
cFLIP expression correlates with tumour progression and patient
outcome in non-Hodgkin lymphomas of low grade of malignancy. Br J
Haematol. 132:560–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeise E, Weichenthal M, Schwarz T and
Kulms D: Resistance of human melanoma cells against the death
ligand TRAIL is reversed by ultraviolet-B radiation via
downregulation of FLIP. J Invest Dermatol. 123:746–754. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ricci MS, Jin Z, Dews M, et al: Direct
repression of FLIP expression by c-myc is a major determinant of
TRAIL sensitivity. Mol Cell Biol. 24:8541–8555. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li W, Zhang X and Olumi AF: MG-132
sensitizes TRAIL-resistant prostate cancer cells by activating
c-Fos/c-Jun heterodimers and repressing c-FLIP(L). Cancer Res.
67:2247–2255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kreuz S, Siegmund D, Rumpf JJ, et al:
NFkappaB activation by Fas is mediated through FADD, caspase-8, and
RIP and is inhibited by FLIP. J Cell Biol. 166:369–380. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Neumann L, Pforr C, Beaudouin J, et al:
Dynamics within the CD95 death-inducing signaling complex decide
life and death of cells. Mol Syst Biol. 6:3522010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu WH, Johnson H and Shu HB: Activation of
NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem.
275:10838–10844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oztürk S, Schleich K and Lavrik IN:
Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners of
life and death decisions. Exp Cell Res. 318:1324–1331.
2012.PubMed/NCBI
|
|
36
|
Amiri KI and Richmond A: Role of nuclear
factor-kappa B in melanoma. Cancer Metastasis Rev. 24:301–313.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mayo MW and Baldwin AS: The transcription
factor NF-kappaB: control of oncogenesis and cancer therapy
resistance. Biochim Biophys Acta. 1470:M55–M62. 2000.PubMed/NCBI
|
|
38
|
Huang S, DeGuzman A, Bucana CD and Fidler
IJ: Nuclear factor-kappaB activity correlates with growth,
angiogenesis, and metastasis of human melanoma cells in nude mice.
Clin Cancer Res. 6:2573–2581. 2000.PubMed/NCBI
|
|
39
|
Galligan L, Longley DB, McEwan M, et al:
Chemotherapy and TRAIL-mediated colon cancer cell death: the roles
of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 4:2026–2036.
2005. View Article : Google Scholar : PubMed/NCBI
|