Introduction
Hepatic cancer is a cancer that originates in the
liver. Liver cancer is a malignant tumor that grows on the surface
or inside of the liver. Several types of liver tumor have been
identified using medical imaging equipment or presented
symptomatically as an abdominal mass, abdominal pain, jaundice,
nausea or liver dysfunction (1,2).
Liver cancer differs from liver metastases, which is a type of
cancer that originates from organs elsewhere in the body and
migrates to the liver. Liver cancer commonly develops resistance to
radiation and chemotherapy and often presents at stages too late
for surgical intervention. Since current treatment modalities are
inadequate, novel therapies are required to treat the various types
of liver cancer, which have an increasing incidence (3). Therefore, identifying targets for
liver cancer is becoming increasingly valuable for the development
of novel methods for therapy.
The endoplasmic reticulum (ER) is an organelle found
in eukaryotic cells that forms an interconnected network of
membrane vesicles (4). The ER is
involved in lipid synthesis, protein folding and maturation and can
be affected by a variety of toxic insults (5,6).
Increasing evidence has verified that ER stress is involved in the
regulation of apoptosis, particularly in ER stress-associated
apoptosis (4–6). ER stress triggers several specific
signaling pathways, including ER-associated protein degradation and
the unfolded protein response (UPR) (4,7,8). The
UPR pathway includes the inositol requiring protein 1 (IRE1)
pathway, the PKR-like ER kinase (PERK) pathway and the activating
transcription factor 6 (ATF6) pathway (9). All the above three UPR pathways are
able to activate the apoptosis associated pro-apoptotic response,
including C/EBP homologous protein (CHOP)/GADD153 and caspase 12,
which finally induces apoptosis.
Leptin is a 16-kDa protein hormone that is important
in regulating energy intake and expenditure, including appetite,
hunger, metabolism and behavior (10). Furthermore, leptin is a pleiotropic
hormone with proliferative and anti-apoptotic roles. Several
studies over the past few years have suggested that leptin/leptin
receptor dysregulation is important in the development of various
types of malignancy, including lung (11), breast (12), gastric (13) and thyroid cancer (14). However, the anti-apoptotic effect
and specific mechanisms underlying the role of leptin in liver
cancer remain to be elucidated. The present study established an
understanding of the anti-apoptotic mechanism of the leptin protein
in liver cancer.
Materials and methods
Cell culture and transfection
The human normal liver cell line HL-7702 and the
hepatocellular carcinoma cell line HepG2 were purchased from ATCC
(Manassas, VA, USA). The two cell lines were maintained in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Carlsbad, CA,
USA) with 15% fetal bovine serum (HyClone, Logan, UT, USA) and
cultured at 37°C with 5% CO2. Different quantities of
plasmids (Invitrogen Life Technologies, Carlsbad, CA, USA) were
transfected into the monolayer cells with Lipofectamine™ 2000
transfection reagent (Invitrogen Life Technologies). The cells were
harvested with trypsin/EDTA (Biyuntian, Beijing, China) in
phosphate-buffered saline (PBS) 24 h post transfection, pelleted by
short centrifugation and suspended in lysis buffer as previously
described (15).
Leptin plasmid construction and leptin
peptide synthesis
The leptin gene was amplified using the polymerase
chain reaction (PCR) technique, using the cDNA of human adipocyte
cells isolated from the subcutaneous fat of a patient (the patient
who provided the subcutaneous fat signed the approved written
informed consent form prior to the present study). The samples were
obtained from The Second Affiliated Hospital of Nanchang
University. The present study was approved by the ethics committee
of The Second Affiliated Hospital of Nanchang University (Nanchang,
China). The primer sequences used were as follows: leptin, forward
5′-GCGAATTCATGGTTCCAATCCAAAAAGTCCAAG AGG-3′ and reverse
5′-TATGGATCCTCAGCACCCAGG GCTGAGG-3′. The PCR product was subcloned
into the pcDNA3.1(+) vector, yielding the recombinant plasmid
pcDNA3.1-ex-LPT. Different quantities of plasmids (2 μg DNA per
well in a 6-well plate and 0.2 μg DNA per well in a 96 plate) were
transfected into the monolayer cells. The other procedures were
performed as described previously (15).
The human leptin peptide was synthesized according
to the following sequence:
ASN-VAL-ILE-GLN-ILE-SER-ASN-ASP-LEU-GLU-ASN-LEU-ARG, pep-LPT
(Sigma, St. Louis, MO, USA). The human leptin peptide (100 nm) was
transfected into BEAS2B cells 4 h prior to treatment with or
without cisplatin.
Small interfering RNA (siRNA)
transfection
siRNA against human leptin (si-LPT) were synthesized
by Invitrogen Life Technologies. Transient transfection was
performed using Lipofectamine™ 2000 transfection reagent according
to the manufacturer’s instructions. HepG2 cells were seeded into
six-well plates for RNA or protein preparation and 96-well plates
for DNA fragmentation or cell growth assays (HepG2-si-LPT).
Following 24 h incubation, the media were replaced with serum-free
RPMI-1640 containing siRNA (100 nmol/l) and transfection reagent.
The experiments were repeated at least three times.
Western blot analysis
All the lysates extracted from the cells were
separated by 15% SDS-PAGE and electro-transferred onto
nitrocellulose membranes. The nitrocellulose membranes were
inhibited with 5% skimmed milk in PBS overnight at 4°C. The
membranes were then incubated with 1:3,000 mouse anti-human
leptin-specific monoclonal antibody (mAb; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), 1:1,000 goat anti-human CHOP pAb
(Stressgen, San Diego, CA, USA), 1:2,000 mouse anti-human p-PERK
mAb (Stressgen), 1:4,000 mouse anti-human ATF6 mAb (Stressgen),
1:3,000 mouse anti-human IRE1 mAb (Santa Cruz Biotechnology, Inc.),
1:2,000 mouse anti-human full length and mouse anti-human spliced
caspase 12 mAb (Stressgen), 1:3,000 anti-human CHOP mAb (Santa Cruz
Biotechnology, Inc.) and 1:1,000 mouse anti-human β-actin mAb
(Santa Cruz Biotechnology, Inc.) for 2 h at room temperature. The
membranes were then incubated with 1:4,000 horseradish
peroxidase-conjugated anti-mouse, 1:1,000 anti-rabbit or anti-goat
immunoglobulin G (Santa Cruz Biotechnology, Inc.). The reactive
signals were visualized using an enhanced chemiluminescence kit (PE
Applied Biosystems, Foster City, CA, USA).
Cell proliferation detection
A
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
(XTT) assay was employed to measure the cell proliferation using a
cytotoxicity detection kit (Sigma). Different quantities of
plasmids (2 μg DNA per well in a 6-well plate and 0.2 μg DNA per
well in a 96 plate) were transfected into the monolayer cells.
Briefly, 24 h after transfection, the cell activity in each well
was detected according to the manufacturer’s instructions. The
96-well plate was read at 490 nm on an ELISA plate reader (MK3;
Thermo Fisher Scientific, Waltham, MA, USA). Each analysis was
performed twice in at least six wells.
Detection of apoptosis
In the present study, flow cytometric analysis, DNA
fragmentation and terminal deoxynucleotidyl transferase dUTP nick
end labeling (TUNEL) assays were employed to detect apoptosis.
Apoptosis was detected by flow cytometric analysis that monitored
annexin V-fluorescein isothiocyanate binding and propidium iodide
uptake simultaneously according to the manufacturer’s instructions
(Sigma). The samples were analyzed by fluorescence on a FACScan
flow cytometer (Beckman Coulter, Miami, FL, USA). DNA fragmentation
in the cells was determined by the DNA-laddering technique as
previously described (15).
Potential DNA fragmentation was examined using the TUNEL apoptosis
detection kit (Chemicon, Temecula, CA, USA).
RNA extraction and semi-quantitative
reverse transcription PCR (RT-PCR)
For mRNA detection of p-Perk, IRE1 and ATF6, a
series of semi-quantitative RT-PCR assays were performed. The
specific primers were synthesized according to previous studies
(Table I) (16–18).
In parallel, individual β-actin was selected as the internal
control. With an RNAsimple Total RNA kit (Tiangen, Beijing, China),
total cellular RNA was prepared. Reverse transcription and RT-PCR
were performed using the SuperScript™ III First-Strand Synthesis
System (Invitrogen Life Technologies) according to the
manufacturer’s instructions. Following electrophoresis on 1.5%
agarose gel, the gel images of each PCR product were digitally
captured with a CCD camera (Canon EOS T3i; Canon, Tokyo, Japan) and
analyzed with the NIH Imager β version 2 (Bio-Rad Gel Documentation
System 2000; Bio-Rad, Hercules, CA, USA). The relative
transcriptional values of each factor in semi-quantitative RT-PCR
are presented as the ratio of the signal value of the specific PCR
product and that of the individual β-actin.
 | Table ISequences of the primers for
endoplasmic reticulum stress-associated genes. |
Table I
Sequences of the primers for
endoplasmic reticulum stress-associated genes.
| Gene | Sequence |
|---|
| β-actin | Forward:
GGACTTCGAGCAGGAGATGG
Reverse: GCACCGTGTTGGCGTAGAGG |
| p-PERK | Forward:
ATCCCCCATGGAACGACCTG
Reverse: ACCCGCCAGGGACAAAAATG |
| IRE1 | Forward:
GAAGACGTCATTGCACGTGAATT
Reverse: AGGTCCTGAATTTACGCAGGT |
| Cleaved | Forward:
GCTTCCAGCAGCACCCAAGAC |
| ATF6 | Reverse:
CGTCTGGCCTTTAGTGGGTGCA |
Statistical analysis
Quantitative analysis of immunoblot images was
performed using the computer-assisted software Image Total Tech
(Pharmacia, St. Paul, MN, USA). Briefly, the image of the
immunoblot was scanned using a Typhoon laser scanner (Typhoon 8600;
Amersham Pharmacia Biotech, Pompano Beach, FL, USA), digitalized
and saved in the TIF format. The average gray value of each
preparation was calculated by the gray numerical value of each blot
vs. that of β-actin. Average data of each preparation were
evaluated from three independent blots and are presented as the
mean ± standard deviation. Statistical analysis was performed using
Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of leptin protein
In the preparations of HepG2 cells, high levels of
intrinsic leptin were detected. The incubation with leptin peptide
(pep-LPT) and expression of leptin (ex-LPT) also generated high
levels of leptin in HL-7702 cells. In si-LPT cells, no significant
leptin bands were detected (Fig. 1A
and B). Furthermore, intrinsically expressed leptin levels in
the HepG2 cells were significantly higher compared with the
incubated or expressed leptin in the pep-LTP and ex-LPT groups
(Fig. 1B; P<0.05).
Leptin expression triggers liver cell
proliferation
XTT analysis revealed that no significant
differences in proliferation and viability among the HepG2, pep-LPT
and ex-LPT groups treated with and without cisplatin were present
(Fig. 1C). However, when the
si-LPT group was treated with cisplatin, the proliferation
viability significantly decreased as compared with the group
treated without cisplatin (Fig.
1C; P<0.001).
Leptin inhibits cell apoptosis
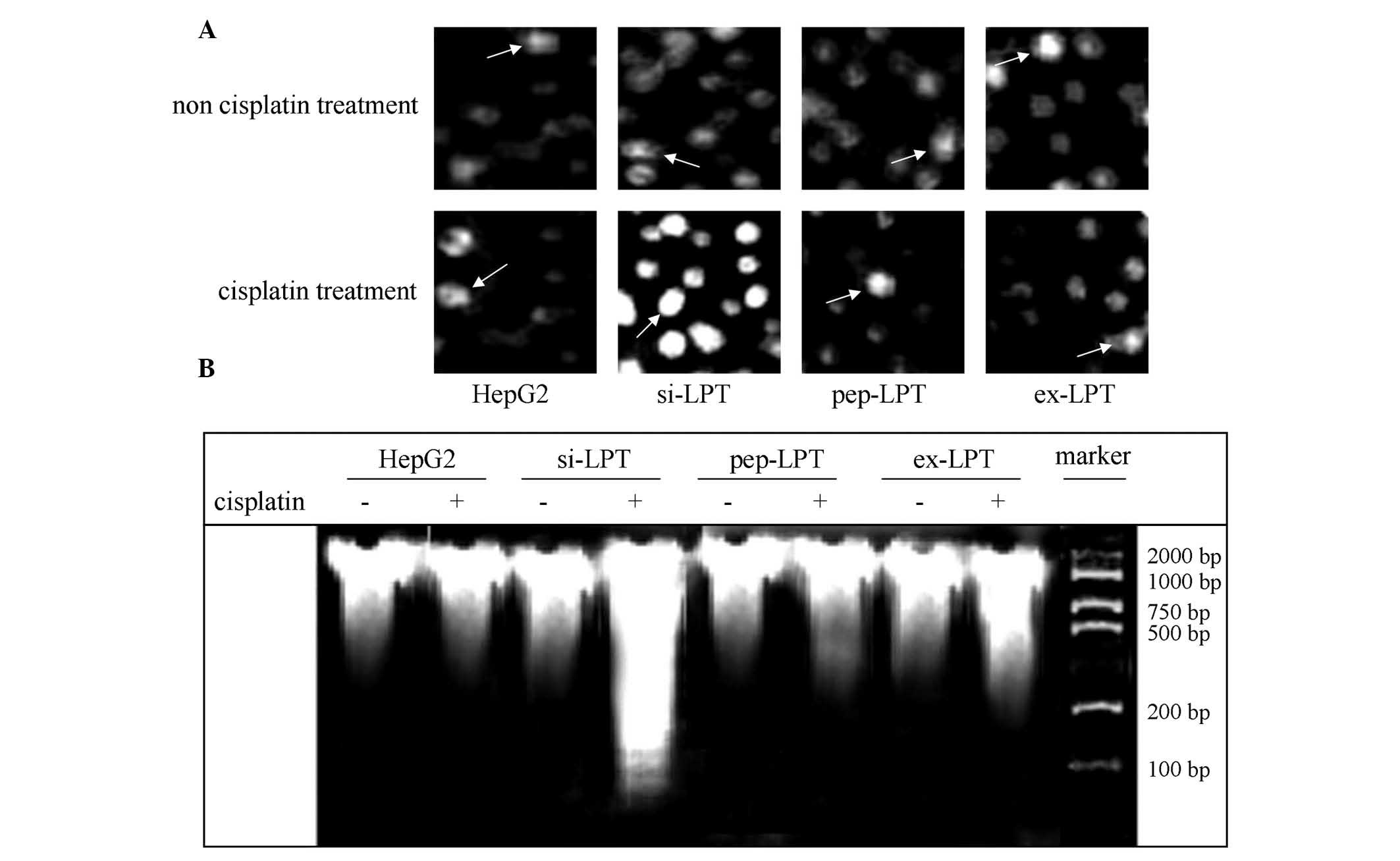
Cell apoptosis was detected by TUNEL and DNA ladder
analysis in the present study. The TUNEL results indicated that
cisplatin was not able to affect the apoptosis of HepG2, pep-LPT
and ex-LPT cells expressing or accumulating leptin protein
(Fig. 2A). In the si-LPT group,
cisplatin treatment was able to significantly induce apoptosis
compared with the group that was not treated with cisplatin
(Fig. 2A). Clear DNA ladders in
agarose electrophoresis gels were observed in the cells in which
the leptin expression was inhibited following treatment with
cisplatin (Fig. 2B). This
indicated that the expression of leptin in normal liver cells was
able to inhibit apoptosis.
The PERK UPR pathway is involved in the
inhibition of leptin-triggered apoptosis
The three main UPR factors of ER stress, PERK, IRE1
and ATF6, were analyzed by western blot analysis and RT-PCR
analysis, respectively. The results demonstrated that the
expression of leptin inhibited the phosphorylation of PERK
(Fig. 3A–C). Therefore, the
quantity of p-PERK was not triggered by the treatment of cisplatin
in the HepG2, pep-PLT and ex-PLT groups (Fig. 3A–C). However, in the si-PLT group,
the phosphorylation of PERK was significantly increased when
treated with cisplatin (Fig. 3D;
P<0.001).
Additionally, the mRNA of the above UPR proteins was
analyzed using semi-quantitative RT-PCR 12 h after transfection. As
shown in Fig. 4, following
treatment with cisplatin, the mRNA levels of p-PERK significantly
increased in the preparations of the si-LPT group as compared with
the groups not treated with cisplatin (Fig. 4C; P<0.05). Notably, no
significant differences in IRE1 and cleaved ATF6 were identified
between the HepG2, pep-LPT and ex-LPT groups in the presence and
absence of cisplatin (Fig. 4A and
B).
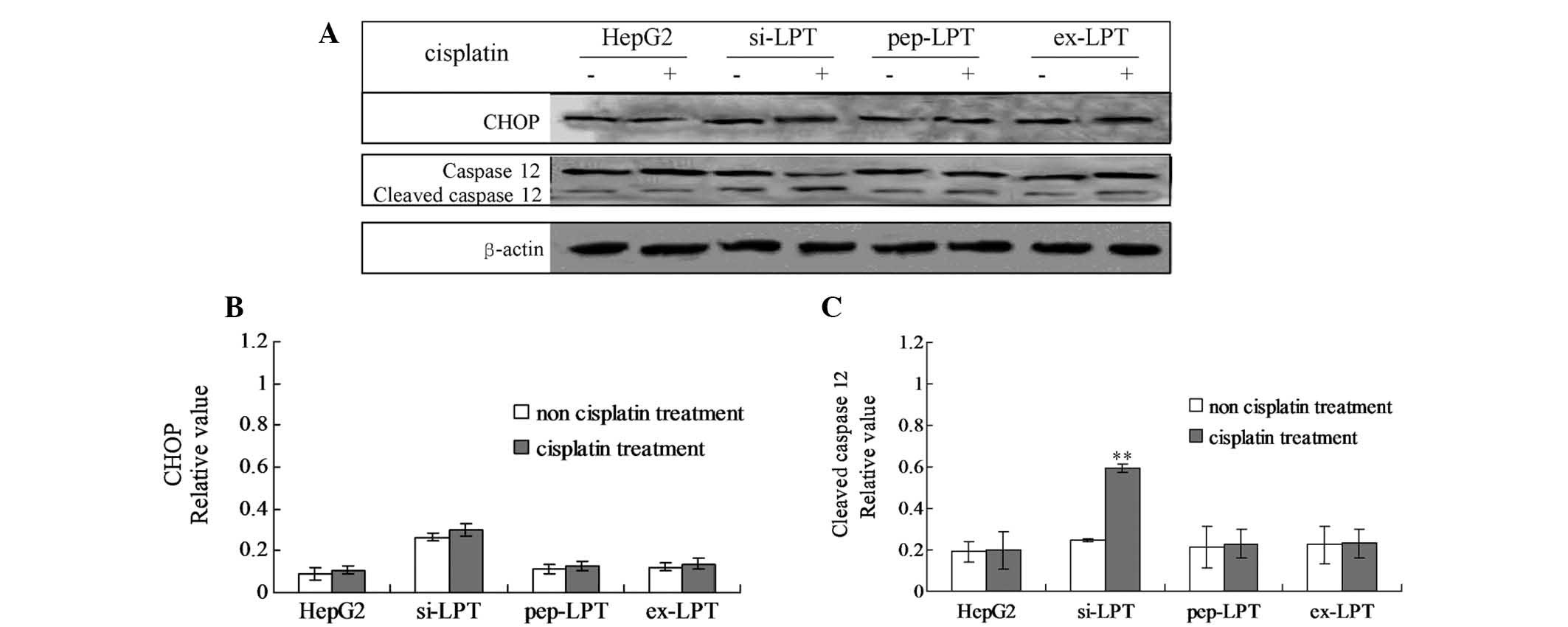
Leptin protein inhibits cleaved caspase
12-induced apoptosis
In order to examine the specific pathway of
leptin-induced inhibition of apoptosis, the cellular levels of
cleaved caspase 12 and the CHOP protein were evaluated by
individual western blot analysis (Fig.
5A). In the cells that were not treated with cisplatin, no
changes in the caspase 12 and CHOP protein were identified in any
of the groups (Fig. 5B and C). No
significant differences were identified in CHOP protein levels in
the cisplatin-treated cells compared with the cells that were not
treated with cisplatin in all the groups (Fig. 5B). Following treatment with
cisplatin, the levels of cleaved caspase 12 in preparations of
si-LPT were significantly higher than those in the groups that were
not treated with cisplatin (Fig.
5C). However, no changes in cleaved caspase 12 were identified
in cells in the presence or absence of cisplatin in the HepG2,
pep-LPT and ex-LPT groups (Fig.
5C). The results indicated that CHOP was activated in the
si-LPT cells and triggered apoptosis.
Discussion
Although a few studies have examined the association
between cancer and the leptin protein (19–22),
the present study is the first one, to the best of our knowledge,
that investigated the specific mechanism underlying the effects of
the leptin protein on apoptosis in the HepG2 cell line. The normal
human liver cell line, HL-7702, was able to evade cisplatin-induced
apoptotic effects when expressing the leptin protein. The present
study demonstrated that leptin inhibits apoptosis through the PERK
pathway and inhibits apoptosis or enhances cell proliferation by
inhibiting the activation of caspase 12.
Levels of intrinsically expressed leptin in HepG2
cells and extrinsically expressed leptin in HL-7702 cells were
examined. The results indicated that all the groups expressed the
leptin protein, however, the levels of leptin in HepG2, pep-LPT and
ex-LPT cells were significantly higher compared with that in the
si-LPT cells. It is known that the malignant transformation of
cancer requires continuous cell growth and the inhibition of
apoptosis. Thus, cell proliferation in the HepG2, pep-LPT, ex-LPT
and si-LPT groups was assessed. The results demonstrated that cell
proliferation was significantly enhanced in the cells expressing
leptin. Therefore, it was hypothesized that the increased cell
proliferation in the HepG2, pep-LPT and ex-LPT cells was due to the
inhibition of apoptosis. A previous study also indicated that
leptin was associated with apoptosis mediated by oxidative stress
(23). In the present study, ER
stress was involved in apoptosis.
In order to investigate the specific mechanism
underlying cell proliferation in leptin-expressing cells, the
levels of ER stress (UPR pathway)-associated proteins, including
p-PERK, IRE1 and cleaved ATF6, were detected. The present study
revealed that the levels of p-PERK protein and mRNA were activated
in the si-LPT group cells following treatment with cisplatin. Thus,
the PERK pathway may be involved in leptin-induced inhibition of
apoptosis. The leptin-induced inhibition of apoptosis may further
elucidate the role of leptin in cancer progression. Previous
studies have demonstrated that the activation of PERK was able to
phosphorylate eukaryotic initiation factor 2, which was able to
activate apoptosis (24). The data
indicated the emergence of ER stress following treatment with
cisplatin in si-LPT cells. Notably, the expression of leptin
inhibited cisplatin-induced ER stress in pep-LPT, ex-LPT and HepG2
cells. Therefore, the present study hypothesized that the leptin
protein may be involved in the pathogenic process of liver
cancer.
ER stress-associated factors (cleaved caspase 12 and
CHOP protein) were detected to identify the specific apoptotic
factors that leptin inhibited. According to a study by Wang et
al (25), cleaved caspase 12
is able to activate caspase 3 and trigger apoptosis, and CHOP can
directly induce ER stress-associated apoptosis. In the present
study, no significant changes in the CHOP protein (activated) were
identified in all the groups (P>0.05) when treated with
cisplatin. Notably, following treatment with cisplatin, cleaved
caspase 12 levels in the si-LPT group were significantly increased
compared with cells that were not treated with cisplatin
(P<0.05); however, no changes in the HepG2, pep-LPT and ex-LPT
groups were identified. The above results, indicate that the leptin
in liver cells was able to indirectly inhibit caspase 12 protein
cleavage, leading to apoptosis. The activation of caspase 12
inhibits cell proliferation by an apoptosis-associated mechanism
(26,27). The present study indicated that
leptin was able to inhibit CHOP-induced ER stress.
In conclusion, the present study suggested that
leptin promotes the growth of HepG2 liver cancer cells through
inhibiting the ER stress-mediated pathway. This inhibition is
triggered by the p-PERK and ATF6 pathway through inhibiting the
expression of CHOP.
Acknowledgements
The present study was supported the Chinese National
Natural Science Foundation (no. 91029720).
References
|
1
|
Wang Y, O’Connor M, Xu Y and Liu X:
Symptom clusters in Chinese patients with primary liver cancer.
Oncol Nurs Forum. 39:E468–E479. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li Y, Huang X, Zhang Q and Ma K:
Phosphorylation of cMet tyrosine residues in murine ascetic hepatic
cancer cell lines with different lymph node metastatic potentials.
Mol Med Rep. 8:655–661. 2013.PubMed/NCBI
|
|
3
|
Yang X, Zu X, Tang J, Xiong W, Zhang Y,
Liu F and Jiang Y: Zbtb7 suppresses the expression of CDK2 and E2F4
in liver cancer cells: implications for the role of Zbtb7 in cell
cycle regulation. Mol Med Rep. 5:1475–1480. 2012.PubMed/NCBI
|
|
4
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moenner M, Pluquet O, Bouchecareilh M and
Chevet E: Integrated endoplasmic reticulum stress responses in
cancer. Cancer Res. 67:10631–10634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Zhang HQ, Zhu GH, Wang YH, Yu XC,
Zhu XB, Liang G, Xiao J and Li XK: A novel mono-carbonyl analogue
of curcumin induces apoptosis in ovarian carcinoma cells via
endoplasmic reticulum stress and reactive oxygen species
production. Mol Med Rep. 5:739–744. 2012.
|
|
7
|
Banjerdpongchai R, Punyati P, Nakrob A,
Pompimon W and Kongtawelert P: 4′-Hydroxycinnamaldehyde from
Alpinia galanga (Linn) induces human leukemic cell apoptosis
via mitochondrial and endoplasmic reticulum stress pathways. Asian
Pac J Cancer Prev. 12:593–598. 2011.
|
|
8
|
Choi EJ and Kim T: Equol induced apoptosis
via cell cycle arrest in human breast cancer MDA-MB-453 but not
MCF-7 cells. Mol Med Rep. 1:239–244. 2008.PubMed/NCBI
|
|
9
|
Hung JY, Hsu YL and Ni WC: Oxidative and
endoplasmic reticulum stress signaling are involved in
dehydrocostuslactone-mediated apoptosis in human non-small cell
lung cancer cells. Lung Cancer. 68:355–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brennan AM and Mantzoros CS: Drug insight:
the role of leptin in human physiology and pathophysiology -
emerging clinical applications. Nat Clin Pract Endocrinol Metab.
2:318–327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duru S, Sönmez Z, Saygideğer Y, Sever O,
Onal B and Ardiç S: The relationship between stage and tumor type
and serum leptin level and leptin expression on tumor tissue in
lung cancer. Tuberk Toraks. 59:427–428. 2011.(In Turkish).
|
|
12
|
Yerlikaya A, Altikat S, Irmak R, Cavga FZ,
Kocacan SA and Boyaci I: Effect of bortezomib in combination with
cisplatin and 5-fluorouracil on 4T1 breast cancer cells. Mol Med
Rep. 8:277–281. 2013.PubMed/NCBI
|
|
13
|
Dong Z, Xu X, Du L, Yang Y, Cheng H, Zhang
X, Li Z, Wang L, Li J, Liu H, Qu X and Wang C: Leptin-mediated
regulation of MT1-MMP localization is KIF1B dependent and enhances
gastric cancer cell invasion. Carcinogenesis. 34:974–983. 2013.
View Article : Google Scholar
|
|
14
|
Zhang GA, Hou S, Han S, Zhou J, Wang X and
Cui W: Clinicopathological implications of leptin and leptin
receptor expression in papillary thyroid cancer. Oncol Lett.
5:797–800. 2013.PubMed/NCBI
|
|
15
|
Wang X, Dong CF, Shi Q, Shi S, Wang GR,
Lei YJ, Xu K, An R, Chen JM, Jiang HY, Tian C, Gao C, Zhao YJ, Han
J and Dong XP: Cytosolic prion protein induces apoptosis in human
neuronal cell SH-SY5Y via mitochondrial disruption pathway. BMB
Rep. 42:444–449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shuda M, Kondoh N, Imazeki N, Tanaka K,
Okada T, Mori K, Hada A, Arai M, Wakatsuki T, Matsubara O, Yamamoto
N and Yamamoto M: Activation of the ATF6, XBP1 and grp78 genes in
human hepatocellular carcinoma: a possible involvement of the ER
stress pathway in hepatocinogenesis. J Hepatol. 38:605–614. 2003.
View Article : Google Scholar
|
|
17
|
Lee do Y, Lee KS, Lee HJ, Kim do H, Noh
YH, Yu K, Jung HY, Lee SH, Lee JY, Youn YC, Jeong Y, Kim DK, Lee WB
and Kim SS: Activation of PERK signaling attenustes Aβ-mediated ER
stress. PLoS One. 5:e104892010.
|
|
18
|
Lee H, Noh JY, Oh Y, Kim Y, Chang JW,
Chung CW, Lee ST, Kim M, Ryu J and Jung YK: IRE1 plays an essential
role in ER stress-mediated aggregation of mutant huntingtin via the
inhibition of autophagy flux. Hum Mol Genet. 21:101–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang S, Zhang Q, Zhang L, Li C and Jiang
H: Expression of ghrelin and leptin during the development of type
2 diabetes mellitus in a rat model. Mol Med Rep. 7:223–228.
2013.PubMed/NCBI
|
|
20
|
Gribovskaja-Rupp I, Kosinski L and Ludwig
KA: Obesity and colorectal cancer. Clin Colon Rectal Surg.
24:229–243. 2011. View Article : Google Scholar
|
|
21
|
Vansaun MN: Molecular pathways:
adiponectin and leptin signaling in cancer. Clin Cancer Res.
19:1926–1932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lai Q and Sun Y: Human leptin protein
induces proliferation of A549 cells via inhibition of PKR-Like ER
kinase and activating transcription factor-6 mediated apoptosis.
Yonsei Med J. 54:1407–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen Y, Wang Q, Zhao Q and Zhou J: Leptin
promotes the immune escape of lung cancer by inducing
proinflammatory cytokines and resistance to apoptosis. Mol Med Rep.
2:295–299. 2009.PubMed/NCBI
|
|
24
|
Kimball SR and Jefferson LS: Induction of
REDD1 gene expression in the liver in response to endoplasmic
reticulum stress is mediated through a PERK, eIF2α phosphorylation,
ATF4-dependent cascade. Biochem Biophys Res Commun. 427:485–489.
2012.PubMed/NCBI
|
|
25
|
Wang X, Shi Q, Xu K, Gao C, Chen C, Li XL,
Wang GR, Tian C, Han J and Dong XP: Familial CJD associated PrP
mutants within the transmembrane region induced Ctm-PrP retention
in ER and trigger apoptosis by the ER stress in SH-SY5Y cells. PLoS
One. 6:e146022011. View Article : Google Scholar
|
|
26
|
Wu CT, Weng TI, Chen LP, Chiang CK and Liu
SH: Involvement of caspase-12-dependent apoptotic pathway in ionic
radiocontrast urografin-induced renal tubular injury. Toxicol Appl
Pharmacol. 266:167–175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang X and Huang N: Berberine induces
selective apoptosis through the AMPK-mediated mitochondrial/caspase
pathway in hepatocellular carcinoma. Mol Med Rep. 8:505–510.
2013.
|