Introduction
Minimal hepatic encephalopathy (MHE) refers to the
subtle changes in cognitive function, electrophysiological
parameters, cerebral neurochemical/neurotransmitter homeostasis,
cerebral blood flow, metabolism and fluid homeostasis that are
observed in patients with cirrhosis, who have no clinical evidence
of hepatic encephalopathy (1). The
glutamate-nitric oxide-cyclic guanine monophosphate (Glu-NO-cGMP)
pathway is involved in certain types of learning and memory
(2,3). Activation of
N-methyl-d-aspartate receptors subunit 1 (NMDAR1) by
glutamate increases calcium in postsynaptic neurons. Calcium binds
to calmodulin (CaM) and activates neuronal nitric oxide synthase
(nNOS), increasing NO, which activates soluble guanylyl cyclase
(sGC), increasing cyclic guanine monophosphate (cGMP), part of
which is released to the extracellular space and is functionally
important in learning and memory (2). A previous study by our group
(4) identified that a
catechol-O-methyltransferase (COMT) inhibitor, a protein involved
in the accumulation of dopamine (DA), was upregulated in cirrhotic
livers in rats with MHE by 2-dimensional gel electrophoresis/mass
spectrometry (2-DE/MS). Furthermore, the levels of DA in cirrhotic
livers, serums and the hippocampi in the MHE group were notably
increased, and the Glu-NO-cGMP memory pathway in hippocampal
neurons was inhibited by the elevation of DA in vivo and
in vitro. Therefore, it was hypothesized that the
pathogenesis of MHE may be associated with the elevation in DA
characteristic of cirrhotic livers. Furthermore, the study
demonstrated that the DA levels were increased in cirrhotic livers
and that it crossed the blood-brain barrier, migrated into the
brains of rats with MHE and inhibited their learning and memory
ability by blocking the Glu-NO-cGMP pathway in neurons (4). However, whether astrocytes were
involved in the pathogenesis of MHE remained elusive.
One type of astrocyte, expressing glutamate
receptors, responds to the synaptic release of glutamate via
channel-mediated currents and is involved in the function of the
Glu-NO-cGMP pathway. Activation of mGluR3 receptors in striatal
neurons (5), hippocampal neurons
(6), cerebellar granule cell
neurons (7) and rat cerebellar
astrocytes (8) is accompanied by a
reduction in the levels of cyclic adenosine monophosphate (cAMP).
In the majority of tissues, NO acts as an intracellular signaling
molecule and is formed in all brain cells, including astrocytes, by
NOS from l-arginine (9). The major
physiological receptor for NO is sGC, an αβ heterodimer that
catalyses the conversion of guanosine triphosphate (GTP) to cGMP
and which acts as a secondary messenger, modulating the activity of
cGMP-dependent protein kinases, cyclic nucleotide
phosphodiesterases and cyclic nucleotide-gated channels (10). Several studies have suggested that
astrocytes integrate learning and memory in the cerebral cortex by
having a direct role in the modulation of synaptic plasticity and
long-term potentiation (11).
The present study delineated the role of the
Glu-NO-cGMP pathway localized in astrocytes in MHE-associated
memory loss, particularly focusing on the effect of elevated DA
from cirrhotic liver in the brains of an MHE in vivo
model.
Materials and methods
MHE models and treatments
A total of 50 Sprague-Dawley rats (Experimental
Animal Center of The Chinese Academy of Sciences in Shanghai,
Shanghai, China) weighing 220–250 g were used. The present study
was approved by the ethics committee of the First Affiliated
Hospital of Wenzhou Medical University (Wenzhou, China) regarding
the care and use of animals for experimental procedures. Rats were
housed under controlled conditions of temperature (24±1°C) and
light (12 h light starting at 07:00 am). Prior to the experimental
stage, all animals were subject to a series of behavioral tests
including Y-maze (YM), open-field (OF), elevated-plus maze (EPM)
and water-finding task (WFT) tests. There was a 15 min interval
between each behavioral test for each rat. The normalized values of
these behavioral tests were obtained. Rats were then randomly
divided into two groups; the control group (n=20) and the
thioacetamide (TAA) group (n=30). MHE was induced by
intraperitoneal injection (i.p.) of TAA (200 mg/kg in normal
saline; Sigma-Aldrich, St. Louis, MO, USA) twice a week for a
period of eight weeks. Then, the rats were subjected to the same
behavioral tests again. Rats included in the MHE group were
required to meet the following criteria: i) The values of one of
the behavioral tests in the MHE group being significantly different
from those of the control group and ii) the EEG revealing no
typical slow wave of hepatic encephalopathy (12). At 24 h following MHE induction,
NMDA (0.3 mM) was also administered to the rats for 30 min by
intraperitoneal injection. Liver, serum and cerebral cortex were
collected for fluorescent staining, immunoblotting and
determination of DA.
DA-injected rat models and
treatments
Rats were administered DA hydrochloride (0.3 and 3
mg/kg; Sigma-Aldrich) by i.p. injection twice per week for four
weeks. All of the rats were subjected to the OF, YM, EPM and WFT
tests. Following the final injection, NMDA (0.3 mM; Sigma-Aldrich)
was also administered to the rats for 30 min by intraperitoneal
injection. Liver, serum and cerebral cortex specimens were
collected for fluorescent staining, immunoblotting and
determination of DA.
Behavioral tests
The OF test was performed as previously described
(13). Briefly, rats were
individually placed at the center of a 10×10 cm gray plastic field
(with 20 cm interval black grids) surrounded by a 20-cm wall and
allowed to move freely for 3 min. Ambulation was measured and
defined as the total number of grid line crossings (13).
The apparatus for the YM test was composed of gray
plastic, with each arm being 40 cm long, 12 cm high, 3 cm wide at
the base and 10 cm wide at the top. The three arms were connected
at an angle of 120°. Rats were individually placed at the end of
one arm and allowed to explore the maze freely for 8 min. Total arm
entries and spontaneous alternation percentage (SA%) were measured.
SA% was defined as the ratio of the arm choices that differed from
the previous two choices (‘successful choices’) to the total
choices during the run (‘total entry minus two’ because the first
two entries were not evaluated). For example, if a mouse made 10
entries, such as 1-2-3-2-3-1-2-3-2-1, there were 5 successful
choices in 8 total choices (10 entries minus 2; 13,14).
The EPM test apparatus was composed of four crossed
arms. Two arms were open (50×10 cm grey plastic floor plate without
walls), whereas the other two were closed (same floor plates with
20 cm-high transparent acrylic wall). The maze was set at a height
of 100 cm above the floor. Rats were allowed to explore the maze
freely for 90 sec. The parameters that were examined were as
follows: (i) The transfer latency (the time elapsed until the first
entry to a closed arm); (ii) the duration of the first stay in a
closed arm (the time from the first entry to a closed arm to the
first escape from the arm) and (iii) the cumulative time spent in
the open/closed arms (13,15).
The WFT test was performed to analyze latent
learning or retention of spatial attention ability in the rats. The
testing apparatus consisted of a grey plastic rectangular open
field (50×30 cm, with a black 10 cm2 grid) with a 15 cm
high wall and a cubic alcove (10×10×10 cm), which was attached to
the center of one longer wall. A drinking tube was inserted through
a hole at the center of the alcove ceiling, with the tip of the
tube placed at 5 cm for training or at 7 cm for the trial from the
floor. A mouse was first placed at the near-right corner of the
apparatus and allowed to explore freely for 3 min. Rats were
excluded from the analysis when they were not able to locate the
tube within the 3 min exploration. Following completion of the
training, the rats were deprived of water for 24 h. In the trial
session, rats were again individually placed at the same corner of
the apparatus and allowed to locate and drink the water in the
alcove. The elapsed time until the first entry into the alcove
(entry latency, EL), until the first touching, sniffing or licking
of the water tube (contacting latency, CL) and until the initiation
of drinking from the water tube (drinking latency, DL) were
measured (13,16,17).
Histopathology
Liver tissues were fixed in 10% formalin for 24 h and
then paraffin-embedded in an automated tissue processor; 5 μm
sections were stained with hematoxylin and eosin (H&E) or
Sirius red and subjected to histopathological examination.
Determination of DA levels
A total of 300–800 μl of 0.4 M HClO4
solution containing 0.1% (w/v)
Na2S2O5 was added to the liver,
serum or cerebral cortex samples, and the mixture was homogenized
by sonication (Labsonic U; B. Braun Biotech International Gmbh,
Melsungen, Germany). The homogenates were centrifuged for 15 min at
20,000 × g at 4°C and aliquots of the supernatants were obtained
for analysis of DA using a high performance liquid chromatography
(HPLC) technique (E2695; Waters, Inc., Milford, MA, USA) (18).
Double-labeled fluorescent staining of
cerebral cortex sections
Four-micron frozen cerebral cortex sections fixed in
acetone or 4% formaldehyde were blocked for endogenous peroxidase
activity with 0.03% H2O2 if appropriate.
Blocking was achieved with phosphate-buffered saline (PBS)
containing 5% normal goat serum (Wuhan Boster Biological
Technology, Ltd., Wuhan, China) for 1 h at room temperature.
Sections were then incubated overnight at 4°C with the following
primary antibodies; NMDAR1 (1:100; mouse monoclonal; Abcam,
Cambridge, MA, USA), CaM (1:100; mouse monoclonal; Abcam), nNOS
(1:50; Rabbit monoclonal; Abcam), sGC (1:50; rabbit polyclonal;
Abcam), cGMP (1:50; mouse monoclonal; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and GFAP (1:50; rabbit polyclonal/mouse
monoclonal; Abcam). Binding of primary antibodies was detected by
incubating the sections for 30 min with fluorescein isothiocyanate
(FITC) (green)/Alexa Fluor 594 (red) conjugated secondary antibody.
Imaging was performed with a Leica TCS SP2 confocal laser scanning
microscope (Leica Microsystems, Wetzlar, Germany). The image data
were analyzed and quantified using ImagePro Plus software 6.0
(Media Cybernetics, Inc., Rockville, MD, USA).
Isolation of astrocytes
Primary cortical astrocytes (PCAs) were prepared
from one-day-old Sprague-Dawley rat pups (19). Tissues of cerebral cortex were
dissociated into a cell suspension using mechanical digestion.
Cells were plated in 75 cm2 tissue culture flasks at a
concentration of 15×106 cells in 11 ml medium and
incubated for 72 h. The medium was changed at this time-point and
every 72 h. Following incubation of the primary cultures for seven
days, the medium was changed completely (11 ml) and the caps were
tightened. Flasks were wrapped in plastic, placed on a shaker
platform in a horizontal position with the medium covering the
cells and centrifuged at 200 × g for 18 h at 37°C to separate the
oligodendrocytes from the astrocytes. The contents were then poured
into a new 75 cm2 flask and incubated for seven days.
Following this, the cells were plated in poly-l-lysine-precoated
six-well plates, incubated with DA (final concentrations of 5 or 50
μM) in 1% serum-containing DMEM/F12 medium for 24 h. Then, 0.3
mmol/l NMDA was added and the incubation continued for another 5
min.
Changes in intracellular Ca2+
in PCAs
The changes in intracellular Ca2+ were
monitored in single PCAs by confocal microscopy using Fluo-3/AM as
previously described (20).
Determination of nitrites and
nitrates
The levels of nitrites and nitrates were measured in
PCAs utilizing the Griess method with nitrate reductase (21). A total of 100 μl of the culture
supernatant was mixed with equal volumes of Griess reagent.
Following 10 min at 20–25°C absorbance was measured at 540 nm.
Determination of cGMP levels
The ELISA assay for the quantitative determination
of cGMP in PCAs was then performed using cGMP fluorescent assay
kits (Molecular Devices Co., Inc., Sunnyvale, CA, USA).
Fluorescent staining of PCAs
PCAs were seeded and cultured on glass coverslips
precoated with 0.01% poly-l-lysine (Sigma-Aldrich) for 1 h.
Following treatment of the cells with DA (final concentration of 5
or 50 μM) for 24 h, they were fixed with 4% paraformaldehyde for 30
min and then treated with 0.1% Triton X-100 for 10 min at room
temperature.
Blocking was achieved with PBS containing 5% normal
goat serum for 1 h at room temperature. Sections were then
incubated overnight at 4°C with the following primary antibodies:
NMDAR1 (1:100; Abcam), CaM (1:100; Abcam), nNOS (1:50; Abcam), sGC
(1:50; Abcam) and cGMP (1:50; Santa Cruz Biotechnology, Inc.).
Binding of primary antibodies was detected by incubating the
sections for 30 min with Alexa Fluor 594 (red) conjugated secondary
antibody. Imaging was performed with a Leica TCS SP2 confocal laser
scanning microscope (Leica Microsystems). The image data were
analyzed and quantified using ImagePro Plus software (Media
Cybernetics, Inc.) (22).
Immunoblotting of PCAs
PCAs were harvested in a lysis buffer [50 mM Tris
HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100 and protease inhibitors
(Sigma-Aldrich)]. The total amount of protein was determined by the
bicinchoninic acid protein assay (Amresco, Solon, OH, USA). Samples
(50 μg protein) were separated by 10% SDS-PAGE and electroblotted
to polyvinylidene fluoride membranes, which were blocked by
incubation in 5% non-fat milk powder dissolved in TBS-T (150 mM
NaCl, 50 mM Tris and 0.05% Tween-20). Following transfer, proteins
were probed using a primary antibody; NMDAR1 (1:1000; Cell
Signaling Technology, Inc.), CaM (1:1000; Abcam), nNOS (1:1500;
Abcam), sGC (1:1000; Abcam) and cGMP (1:200, Santa Cruz
Biotechnology). Then, horseradish peroxidase-conjugated secondary
antibody was used. Following extensive washing, protein bands
detected by antibodies were visualized by enhanced
chemiluminescence reagent (Pierce Biotechnology, Inc., Rockford,
IL, USA) following exposure on Kodak BioMax film (Kodak, Rochester,
NY, USA). The films were subsequently scanned and the band
intensities were quantified using Quantity One software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
A two-tailed Student’s t-test was used to determine
the statistical significance of difference in values between the
control and experimental preparations. All data are presented as
the mean ± standard deviation. P<0.05 or P<0.01 were
considered to indicate a statistically significant difference
between values.
Results
Memory impairment and elevation of
intracranial DA levels in MHE models
H&E and Sirius red staining in the livers of
TAA-treated rats revealed inflammatory cell infiltration around the
portal area, with collagen deposition or fibrous septa formation
(Fig. 1A and B), suggesting that
the liver fibrosis model was successfully established.
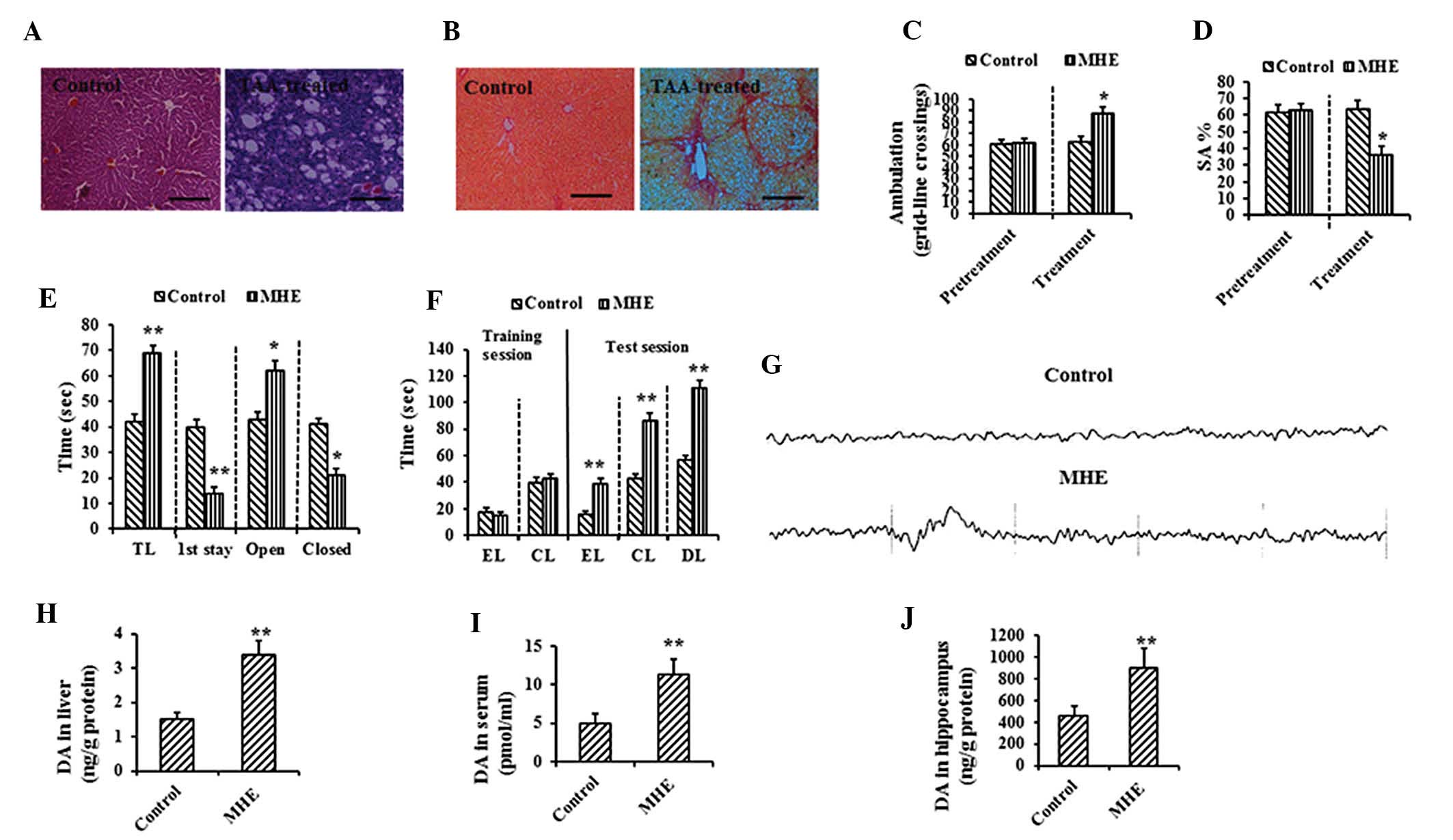 | Figure 1Memory impairment and elevated DA in
MHE models. (A) Hematoxylin and eosin (scale bar, 50 μm) and (B)
Sirius red staining (scale bar, 100 μm) of liver sections from the
control and TAA-treated rats. (C) Ambulation in open-field test of
the control and TAA-treated rats (left, pretreatment; right,
eight-week treatment). (D) SA% in YM of the control and TAA-treated
rats (left, pretreatment; right, 8-week treatment). (E) Results of
EPM (TL, transfer latency; first stay, duration of the first stay;
open, time spent in the open arms; closed, time spent in the closed
arms). (F) Results of the WFT test (EL, entry latency; CL,
contacting latency; DL, drinking latency). (G) The cerebral signal
of rats observed in the scalp EEG falls in the Alpha (8–13 Hz)
range in the control and MHE rats. Levels of DA in (H) the liver,
(I) serum and (J) cerebral cortex analyzed in the control and MHE
groups. Data are presented as the mean ± standard deviation.
*P<0.05, **P<0.01 vs. control treatment
by Dunnett’s post-hoc test. DA, dopamine; MHE, minimal hepatic
encephalopathy; YM, Y-maze; SA%, spontaneous alternation
percentage; EPM, elevated-plus maze; WFT, water-finding task; TAA,
thioacetamide. |
Rats were then subjected to a series of behavioral
tests, including OF, YM, EPM and WFT tests. There were significant
differences in the voluntary activities in the OF test between the
MHE and control groups following treatment (Fig. 1C). The SA% in the YM of the
TAA-treated rats was significantly decreased (P<0.01), compared
with that of the control rats (Fig.
1D). In the EPM test, TAA-treated rats remained in the open
arms for significantly longer than in the closed arms, as compared
with the controls (Fig. 1E). In
the WFT test, significant delays in EL, CL and DL were detected in
the TAA-treated rats as compared with the controls (Fig. 1F). In the EEG tests, 6/25
TAA-treated rats exhibited slow wave (Theta =4–7 Hz or Delta <4
Hz wave; Fig. 1G). Therefore, the
incidence of MHE in the TAA group was 76.0% (19/25).
Considering that the increased levels of DA in the
liver (Fig. 1H), serum (Fig. 1I) and cerebral cortex (Fig. 1J) of MHE rats were observed in a
previous study by our group (4),
the present study examined whether the concentration of DA in the
liver, serum and cerebral cortex of rats with MHE would exhibit
similar trends. The data were consistent with those of the previous
study, confirming the earlier results (Fig. 1H). The results confirmed that DA,
when elevated in the liver, crossed the blood-brain barrier and
permeated into the brains of rats with MHE, as previously
observed.
Confirmation of memory impairment caused
by elevation of intracranial DA
To confirm whether memory impairment in rats with
MHE was associated with DA transported into the brain, normal rats
were subjected to an i.p. injection of DA (low dose, 0.3 mg/kg and
high dose, 3 mg/kg). At a week following the injection, all rats
were again subjected to behavioral tests, including OF, YM, EPM and
WFT tests. The mean voluntary activities in the OF test were
significantly increased following high-dose DA treatment (Fig. 2A). The SA% (P<0.01) in the YM
test was significantly decreased in high-dose DA-treated rats
(Fig. 2B). In the EPM test, the
high-dose DA-treated rats remained in the open arms for
significantly longer and spent a shorter time in the closed arms as
compared with the controls (Fig.
2C). In the WFT, EL, CL and DL tests, the responses were
significantly delayed in the high-dose DA-treated rats as compared
with the controls (Fig. 2C). The
control and DA-treated rats demonstrated an Alpha (8–13 Hz) band in
the EEG tests (Fig. 2E).
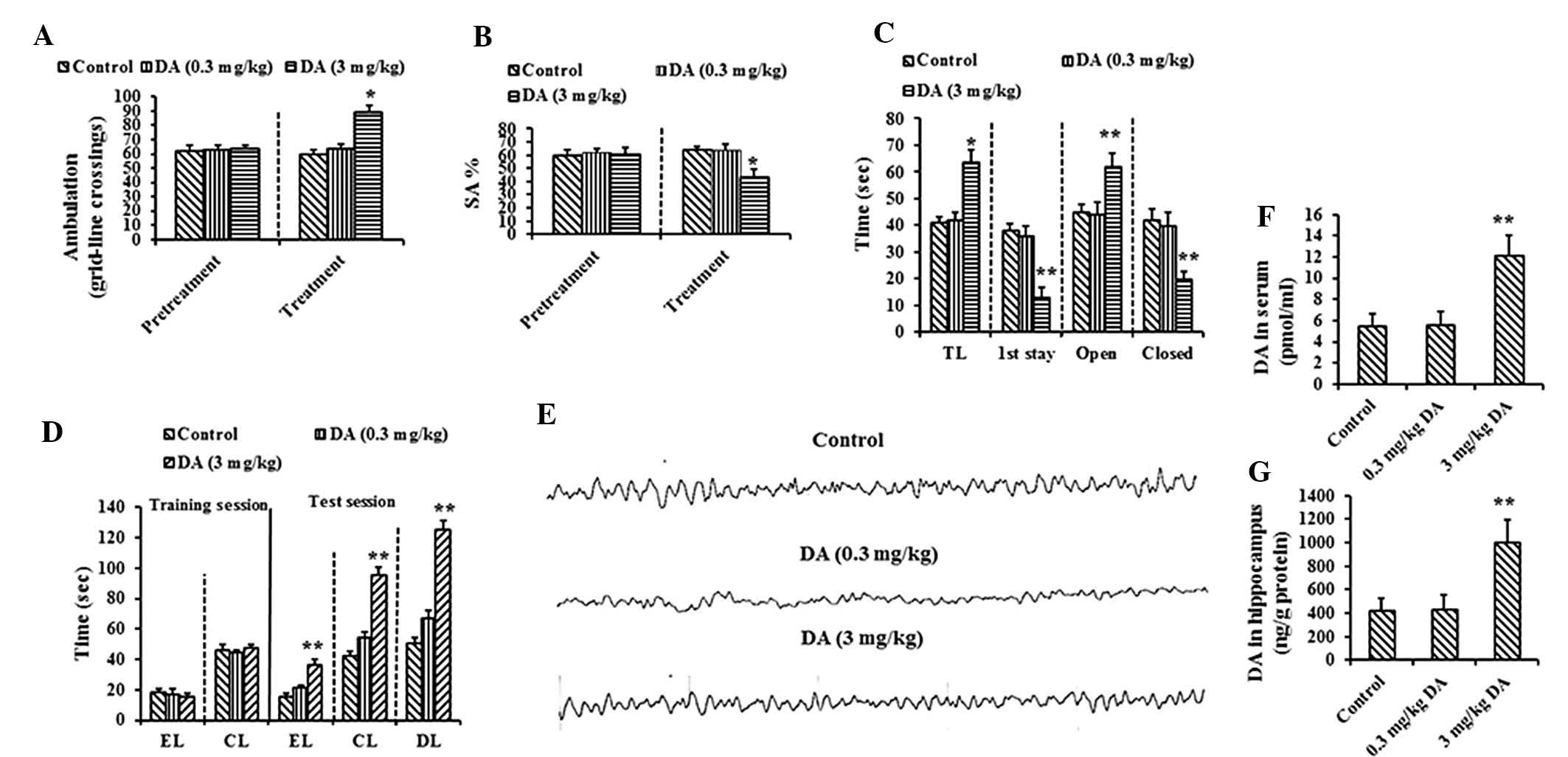 | Figure 2Effect of intracranial elevated DA on
memory impairment. (A) Ambulation in OF of control and DA (0.3 and
3 mg/kg)-treated rats (left, pretreatment; right, eight-week
treatment). (B) SA% in YM of control and DA (0.3 and 3 mg/kg)
-treated rats (left, pretreatment; right, eight-week treatment).
(C) Results of EPM (TL, transfer latency; first stay, duration of
the first stay; open, time spent in the open arms; closed, time
spent in the closed arms). (D) Results of WFT (EL, entry latency;
CL, contacting latency; DL, drinking latency). (E) No Theta (4–7
Hz) or Delta (<4 Hz) bands (slow wave) were observed in EEG of
DA-treated rats. Levels of DA in the (F) serum and (G) cerebral
cortex analyzed in the control and DA-treated groups. Data are
presented as the mean ± standard deviation. *P<0.05,
**P<0.01 vs. control treatment by Dunnett’s post-hoc
test. DA, dopamine; MHE, minimal hepatic encephalopathy; OF,
open-field; YM, Y-maze; SA%, spontaneous alternation percentage;
EPM, elevated-plus maze; WFT, water-finding task; EEG,
electroencephalogram. |
It was also observed that the levels of DA in the
serum (Fig. 2F) and cerebral
cortex (Fig. 2G) were
significantly elevated following treatment with a high dose of DA
(P<0.01), confirming that when blood levels were high, DA
crossed the blood-brain barrier, permeated the brains and
subsequently attenuated cognitive function in rats.
Inactivation of Glu-NO-cGMP pathway in
astrocytes of the cerebral cortex by DA in vivo
A previous study by our group identified that memory
impairment in rats with MHE was associated with the inactivation of
the Glu-NO-cGMP pathway in neurons by high doses of DA in the brain
(4). Therefore, the present study
examined whether the inactivation of the Glu-NO-cGMP pathway in
astrocytes contributed to memory impairment in rats with MHE. The
proteins of the Glu-NO-cGMP pathway (NMDAR1, CaM, nNOS, sGC and
cGMP) were co-localized with GFAP in the cerebral cortex. The
co-localization indicated that the four proteins were significantly
decreased in the rats with MHE, as compared with the normal rats
(Fig. 3A–E), indicating that the
inhibition of the Glu-NO-cGMP pathway in astrocytes of rats with
MHE was associated with memory impairment. As expected, the NMDAR1,
CaM, nNOS, sGC and cGMP levels were substantially increased
following i.p. injection of NMDA (Fig.
3E). The augmentation of NMDAR1, CaM, nNOS, sGC and cGMP levels
induced by NMDA was inhibited in rats with MHE (Fig. 3A–E).
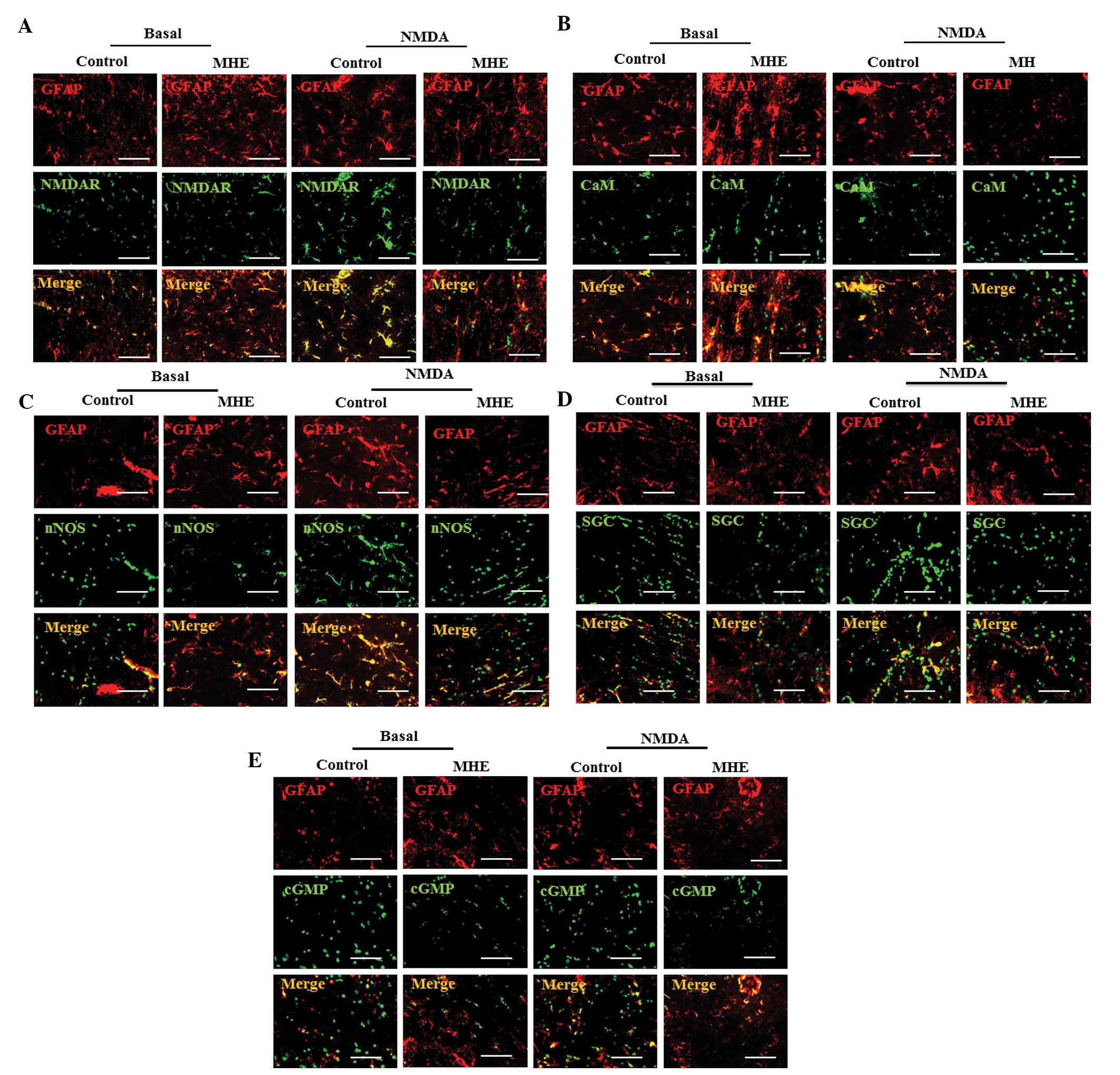 | Figure 3The Glu-NO-cGMP pathway was
inactivated in astrocytes of cerebral cortex in MHE models. (A)
Co-localization of NMDAR1, (B) CaM, (C) nNOS, (D) sGC, (E) cGMP
(green) with GFAP (an astrocytic marker, red) is indicated by the
overlap of signals resulting in yellow staining in the cerebral
cortex of the control and MHE rats with the absence or the presence
of NMDA (0.3 mM) (scale bar, 25 μm). Glu-NO-cGMP, glutamate-nitric
oxide-cyclic guanosine monophosphate; MHE, minimal hepatic
encephalopathy; NMDAR1, N-methyl-d-aspartate receptors
subunit 1; CaM, calmodulin; nNOS, nitric oxide synthase; sGC,
soluble guanylyl cyclase; cGMP, cyclic guanine monophosphate; GFAP,
glial fibrillary acidic protein. |
Following this, it was determined whether DA was
involved in the inhibition of the Glu-NO-cGMP pathway in astrocytes
in the DA (0.3 and 3 mg/kg)-treated rats. DA treatment also induced
a dose-dependent decrease in the expression of the four proteins
localized in the astrocytes of the cerebral cortex of rats. The
co-localization demonstrated that the four proteins were highly
expressed in the astrocytes of normal and low-dose DA-treated rats
and weakly expressed in the astrocytes of high-dose DA-treated rats
(Fig. 4A–E), indicating high-dose
DA-induced inhibition of the Glu-NO-cGMP pathway in the astrocytes
of the cerebral cortex of rats. Furthermore, it was identified that
high-dose DA (3 mg/kg) decreased NMDA-mediated augmentation of
NMDAR1, CaM, nNOS, sGC and cGMP expression levels in astrocytes
(Fig. 4A–E).
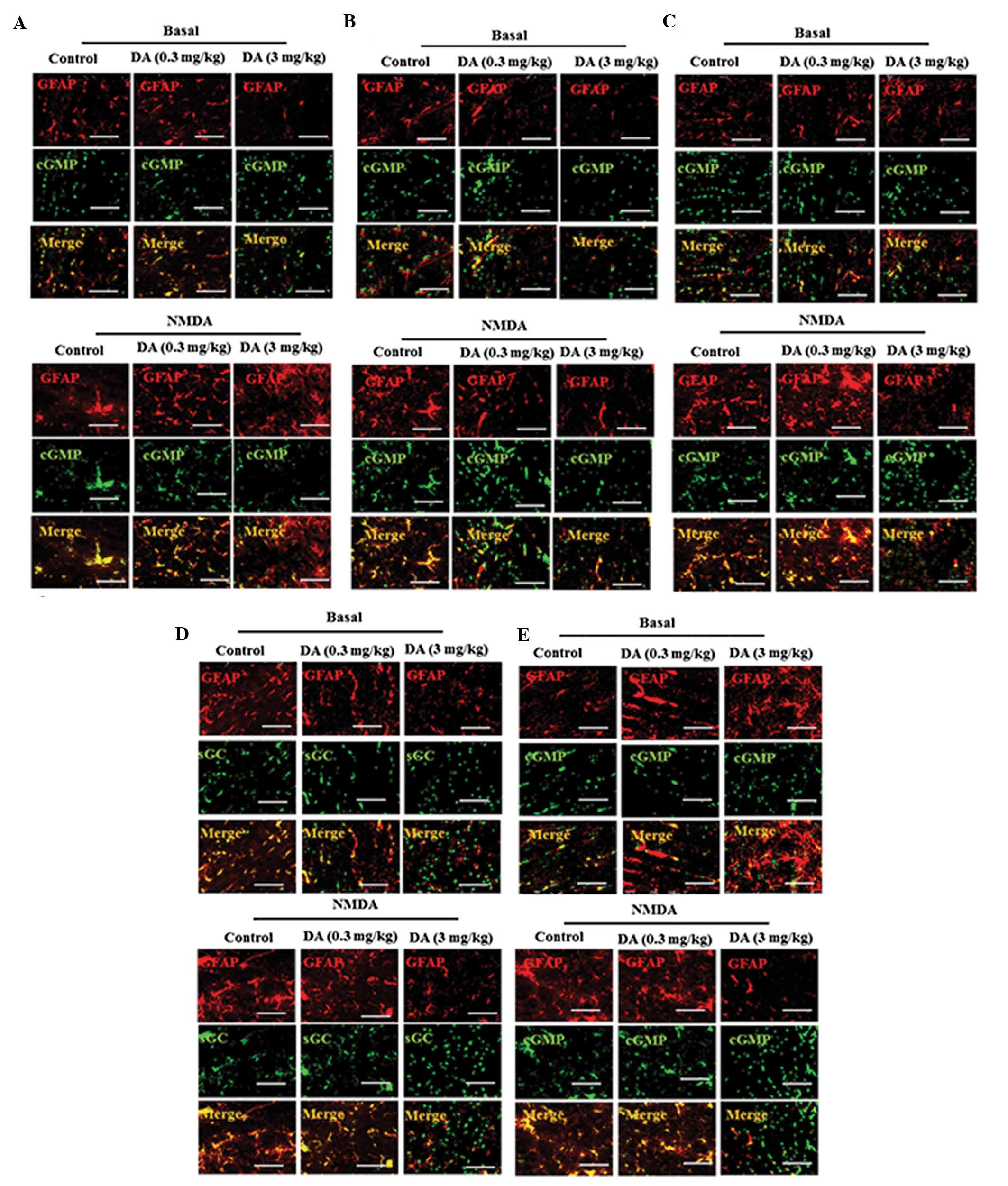 | Figure 4The Glu-NO-cGMP pathway was inhibited
by DA (0.3 and 3 mg/kg) in astrocytes of the cerebral cortex in
vivo. Double immunofluorescent staining of cerebral cortex of
control and DA (0.3 and 3 mg/kg) -treated rats with the absence or
the presence of NMDA using antibodies against GFAP (red) and (A)
NMDAR1, (B) CaM, (C) nNOS, (D) sGC and (E) cGMP (green) (scale bar,
25 μm). Glu-NO-cGMP, glutamate-nitric oxide-cyclic guanosine
monophosphate; NMDAR1, N-methyl-d-aspartate receptors
subunit 1; DA, dopamine; CaM, calmodulin; nNOS, nitric oxide
synthase; sGC, soluble guanylyl cyclase; cGMP, cyclic guanine
monophosphate; GFAP, glial fibrillary acidic protein. |
Inactivation of the Glu-NO-cGMP pathway
in PCAs by DA
To confirm the inhibition of the activation of the
Glu-NO-cGMP pathway by DA in PCAs, the levels of intracellular
Ca2+, nitrites and nitrates and cGMP in DA-treated PCAs
were assessed. NMDA (0.3 mmol/l) was added to the normal and
DA-treated PCAs. As revealed in Fig.
5Aa, NMDA induced increases in calcium levels in normal PCAs.
Chronic exposure to high-dose DA significantly decreased
NMDA-induced increases in calcium (Fig. 5Ad). Basal concentrations of
nitrites and nitrates were lower in high-dose DA-treated PCAs
(1.15±0.89 mol/l) than in the controls (2.51±0.56 mol/l) (Fig. 5B). NMDA increased the levels of
nitrites and nitrates in the control PCAs. NMDA-induced elevation
of NO was also impaired in DA-treated PCAs (Fig. 5B).
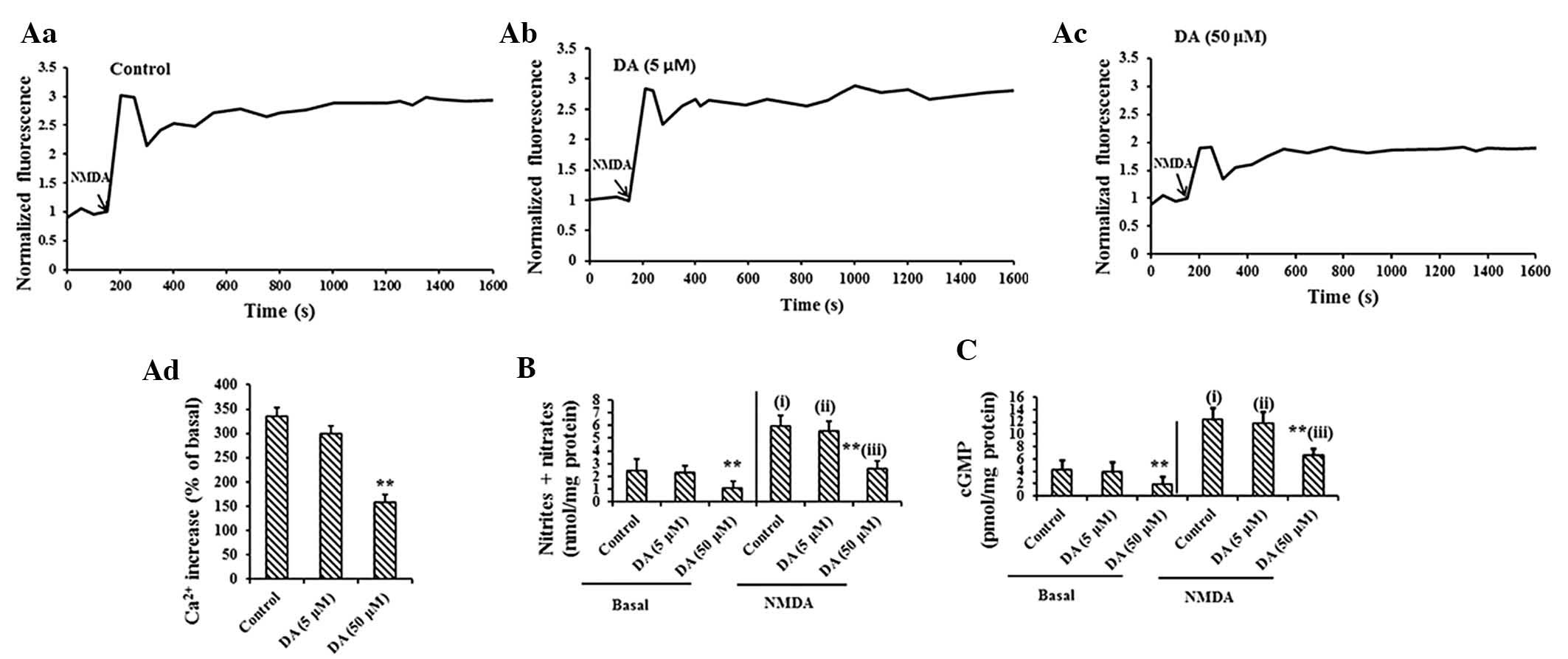 | Figure 5The function of the Glu-NO-cGMP
pathway is impaired in PCAs chronically exposed to DA. (A) Free
intracellular Ca2+ content was followed with fluo-3/AM
using confocal microscopy. The basal Ca2+ levels were
recorded for 200 sec, then 0.3 mM NMDA was added and the
fluorescence was recorded for 1,600 sec. Typical traces are
demonstrated in (Aa), (Ab) and (Ac). The increase in calcium at
1,600 sec compared with 200 sec was similar in (Aa) the control
astrocytes or (Ab) DA-treated astrocytes. The values (mean ± SD) of
quadruplicate samples from six different cultures are illustrated
in (Ad), **P<0.01 vs. controls. (B) The levels of
nitrites under basal conditions, 5 min following the addition of
0.3 mM NMDA. The values were significantly different (P<0.05)
from basal nitrites prior to the addition of NMDA, and are
indicated by (i) for control astrocytes and by (ii) and (iii) for 5
μM and 50 μM DA-treated astrocytes, respectively. Values that are
significantly different in astrocytes exposed to ammonia from the
control astrocytes are indicated by asterisks,
**P<0.01 vs. controls. (C) The content of cGMP in
astrocytes treated or not (basal) with NMDA (0.3 mM) was measured.
Values are the presented as the mean ± SD of triplicate samples
from seven different cultures. Values that were significantly
different (P<0.05) from basal cGMP prior to the addition of NMDA
are indicated by (i) for the control astrocytes and by (ii) and
(iii) for the astrocytes exposed to 5 μM DA and 50 μM DA,
respectively. Values that are significantly different in astrocytes
exposed to DA from control astrocytes are indicated by asterisks,
**P<0.01 vs. controls. PCAs, primary cortical
astrocytes; DA, dopamine; Glu-NO-cGMP, glutamate-nitric
oxide-cyclic guanosine monophosphate; NMDA,
N-methyl-d-aspartate; cGMP, cyclic guanine monophosphate;
SD, standard deviation. |
DA treatment induced a dose-dependent decrease in
the formation of cGMP in PCAs. As shown in Fig. 5C, the content of basal cGMP in PCAs
following low-dose DA treatment (5 μM) revealed no difference
compared with those of the controls. Chronic exposure to high
levels of DA significantly decreased the basal cGMP concentration.
The addition of NMDA increased cGMP levels in the control PCAs, 5
and 50 μM DA-treated PCAs to 12.31±2.02, 11.79±1.84 and 6.53±1.12
pmol/mg protein, respectively (Fig.
5C), indicating a significant reduction of 38% for 50 μM DA
treatment in the function of the Glu-NO-cGMP pathway.
Following this, the effect of DA on the expression
of proteins of the Glu-NO-cGMP pathway was confirmed by
immunostaining and immunoblot analysis of NMDAR1, CaM, nNOS, sGC
and cGMP in PCAs. DA treatment induced a dose-dependent decrease in
the expression of the four proteins in PCAs. The results of the
immunoblotting analyses revealed that the expression of the four
proteins was downregulated in PCAs exposed to high-dose DA (50 μM)
as compared with the control group (Fig. 6A). As demonstrated in Fig. 6B, immunofluorescent assessment
revealed that low-dose DA treatment (5 μM) failed to attenuate the
number of NMDAR1, CaM, nNOS or sGC immunoreactive PCAs. By
contrast, the high-dose DA treatment (50 μM) successfully decreased
the expression of the four proteins. The addition of NMDA
upregulated the expression of the four proteins in PCAs, which was
an effect that was inhibited by high-dose DA (Fig. 6A and B). These results suggested
that DA inhibits the function of the whole Glu-NO-cGMP pathway.
 | Figure 6The effect of DA (5 and 50 μM) on the
Glu-NO-cGMP pathway in PCAs. (A) Immunoblot analysis of PCAs
treated with DA (5 and 50 μM). Expression of NMDAR1, CaM, nNOS, sGC
and cGMP was normalized to the corresponding GAPDH protein. Data
are presented as the mean ± standard deviation. The values were
signifcantly different (P<0.05) from basal nitrites prior to the
addition of NMDA and are indicated by (i) for control astrocytes
and by (ii) and (iii) for 5 μM and 50 μM DA-treated astrocytes,
respectively. *P<0.05, **P<0.01 vs. the
control group. (B) Immunofluorescence staining of DA (5 and 50
μM)-treated PCAs in the absence or the presence of NMDA (0.3 mM)
using the anti-NMDAR1, CaM, nNOS, sGC and cGMP antibodies (scale
bar, 50 μm). PCAs, primary cortical astrocytes; DA, dopamine;
NMDAR1, N-methyl-d-aspartate receptors subunit 1;
Glu-NO-cGMP, glutamate-nitric oxide-cyclic guanosine monophosphate;
CaM, calmodulin; nNOS, nitric oxide synthase; sGC, soluble guanylyl
cyclase; cGMP, cyclic guanine monophosphate. |
Discussion
In the present study, it was demonstrated that
chronic stimulation of astrocytes in the cerebral cortex by toxic
DA significantly deteriorated the expression of proteins involved
in the Glu-NO-cGMP pathway. The results revealed that chronic
exposure to DA affected the Glu-NO-cGMP pathway in astrocytes of
the rat cerebral cortex in vivo at different stages.
Exposure to DA significantly reduced the levels of NMDAR1, CaM,
nNOS, sGC and cGMP (Fig. 3) and
the activation of NMDAR1, CaM, nNOS, sGC and cGMP by NMDA (Fig. 1). This effect may subsequently
contribute to the reduced formation of cGMP. Therefore, the present
study provided the first evidence, to the best of our knowledge,
suggesting that the Glu-NO-cGMP pathway localized in astrocytes may
be important in the pathogenesis of DA-associated memory impairment
in rats with MHE.
Chronic exposure of rats to DA appeared to affect
the Glu-NO-cGMP pathway at different stages. A number of studies
have suggested that high concentrations of DA reduce
NMDAR1-mediated currents and postsynaptic potentials in pyramidal
neurons (23–25). Another recent study reported that
the direct inhibition of NMDAR1 by D1 ligands (DA) is due to the
blockade of the channel pore (26). Several previous studies have
demonstrated that DA inhibits NMDA receptor-mediated nNOS
stimulation, and thus has a role in nNOS-NO signaling in the
generation of striatal cGMP (27–33).
NO is partially controlled by nigrostriatal DA input and affects
striatal functions (34–37). One study suggested that the
parkinsonian state is associated with an abnormal NO/sGC cascade.
The DA-deprived striatum revealed a reduction in the standard
NO-mediated inhibition (38). The
present study demonstrated that the decreased content of NMDAR1,
CaM, nNOS, sGC and cGMP, and the impairment of NMDA-induced
activation of NMDAR1, CaM, nNOS, sGC and cGMP occurred in
astrocytes treated with high concentrations of DA, suggesting that
DA was responsible for impaired activation of any of the proteins
of the Glu-NO-cGMP pathway and was involved in the impairment of
cognitive ability.
In conclusion, the present study provided evidence
for a novel theory accounting for memory dysfunction in MHE. The
results demonstrated that high DA levels in cirrhotic livers led to
elevated DA levels in the brains of MHE models, and that the
subsequent DA-dependent inactivation of the Glu-NO-cGMP pathway in
astrocytes triggered memory impairment in the rats with MHE. The
effect of DA on the impairment of the Glu-NO-cGMP pathway in
astrocytes provided new insights that facilitate the understanding
of the function of astrocytes. These results provided evidence of
not only a novel pathological hallmark of MHE but also a candidate
target for MHE therapy. Further investigations should focus on the
downstream cascades of the Glu-NO-cGMP pathway that may also be
involved in DA-induced memory impairment in MHE.
Acknowledgements
This study was funded by the Natural Science
Foundation of China (81300308 and 81301014).
References
|
1
|
Tan HH, Lee GH, Thia KT, Ng HS, Chow WC
and Lui HF: Minimal hepatic encephalopathy runs a fluctuating
course: results from a three-year prospective cohort follow-up
study. Singapore Med J. 50:255–260. 2009.PubMed/NCBI
|
|
2
|
Yamada K, Hiramatsu M, Noda Y, et al: Role
of nitric oxide and cyclic GMP in the dizocilpine-induced
impairment of spontaneous alternation behavior in mice.
Neuroscience. 74:365–374. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Erceg S, Monfort P, Hernandez-Viadel M,
Rodrigo R, Montoliu C and Felipo V: Oral administration of
sildenafil restores learning ability in rats with hyperammonemia
and with portacaval shunts. Hepatology. 41:299–306. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ding S, Liu L, Jing H, et al: Dopamine
from cirrhotic liver contributes to the impaired learning and
memory ability of hippocampus in minimal hepatic encephalopathy.
Hepatol Int. 7:923–936. 2013. View Article : Google Scholar
|
|
5
|
Manzoni O, Prezeau L, Sladeczek F and
Bockaert J: Trans-ACPD inhibits cAMP formation via a pertussis
toxin-sensitive G-protein. Eur J Pharmacol. 225:357–358. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schoepp DD, Johnson BG and Monn JA:
Inhibition of cyclic AMP formation by a selective metabotropic
glutamate receptor agonist. J Neurochem. 58:1184–1186. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wroblewska B, Wroblewski JT, Saab OH and
Neale JH: N-acetylaspartylglutamate inhibits forskolin-stimulated
cyclic AMP levels via a metabotropic glutamate receptor in cultured
cerebellar granule cells. J Neurochem. 61:943–948. 1993. View Article : Google Scholar
|
|
8
|
Wroblewska B, Santi MR and Neale JH:
N-acetylaspartylglutamate activates cyclic AMP-coupled metabotropic
glutamate receptors in cerebellar astrocytes. Glia. 24:172–179.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bredt DS and Snyder SH: Nitric oxide: a
physiologic messenger molecule. Annu Rev Biochem. 63:175–195. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bellamy TC and Garthwaite J: The
receptor-like properties of nitric oxide-activated soluble guanylyl
cyclase in intact cells. Mol Cell Biochem. 230:165–176. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Newman EA: Propagation of intercellular
calcium waves in retinal astrocytes and Müller cells. J Neurosci.
21:2215–2223. 2001.PubMed/NCBI
|
|
12
|
Jia L and Zhang MH: Comparison of
probiotics and lactulose in the treatment of minimal hepatic
encephalopathy in rats. World J Gastroenterol. 11:908–911. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kawasumi M, Chiba T, Yamada M, et al:
Targeted introduction of V642I mutation in amyloid precursor
protein gene causes functional abnormality resembling early stage
of Alzheimer’s disease in aged mice. Eur J Neurosci. 19:2826–2838.
2004.PubMed/NCBI
|
|
14
|
Yamada M, Chiba T, Sasabe J, et al:
Implanted cannula-mediated repetitive administration of Abeta25–35
into the mouse cerebral ventricle effectively impairs spatial
working memory. Behav Brain Res. 164:139–146. 2005.PubMed/NCBI
|
|
15
|
Itoh J, Nabeshima T and Kameyama T:
Utility of an elevated plus-maze for the evaluation of memory in
mice: effects of nootropics, scopolamine and electroconvulsive
shock. Psychopharmacology (Berl). 101:27–33. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ichihara K, Nabeshima T and Kameyama T:
Differential effects of pimozide and SCH 23390 on acquisition of
learning in mice. Eur J Pharmacol. 164:189–195. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mamiya T, Noda Y, Nishi M, Takeshima H and
Nabeshima T: Enhancement of spatial attention in
nociceptin/orphanin FQ receptor-knockout mice. Brain Res.
783:236–240. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Colado MI, Ormazabal MJ, Alfaro MJ and
Martin MI: Effect of Bay K 8644 on the synthesis and metabolism of
dopamine and 5-hydroxytryptamine in various brain areas of the rat.
J Pharm Pharmacol. 45:220–222. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bernabeu R, Schmitz P, Faillace MP,
Izquierdo I and Medina JH: Hippocampal cGMP and cAMP are
differentially involved in memory processing of inhibitory
avoidance learning. Neuroreport. 7:585–588. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marcaida G, Miñana MD, Burgal M, Grisolía
S and Felipo V: Ammonia prevents activation of NMDA receptors by
glutamate in rat cerebellar neuronal cultures. Eur J Neurosci.
7:2389–2396. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Verdon CP, Burton BA and Prior RL: Sample
pretreatment with nitrate reductase and glucose-6-phosphate
dehydrogenase quantitatively reduces nitrate while avoiding
interference by NADP+ when the Griess reaction is used to assay for
nitrite. Anal Biochem. 224:502–508. 1995. View Article : Google Scholar
|
|
22
|
Yi Z, Petralia RS, Fu Z, et al: The role
of the PDZ protein GIPC in regulating NMDA receptor trafficking. J
Neurosci. 27:11663–11675. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Castro NG, de Mello MC, de Mello FG and
Aracava Y: Direct inhibition of the N-methyl-D-aspartate receptor
channel by dopamine and (+)-SKF38393. Br J Pharmacol.
126:1847–1855. 1999. View Article : Google Scholar
|
|
24
|
Zheng P, Zhang XX, Bunney BS and Shi WX:
Opposite modulation of cortical N-methyl-D-aspartate
receptor-mediated responses by low and high concentrations of
dopamine. Neuroscience. 91:527–535. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Seamans JK and Yang CR: The principal
features and mechanisms of dopamine modulation in the prefrontal
cortex. Prog Neurobiol. 74:1–58. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cui C, Xu M and Atzori M:
Voltage-dependent block of N-methyl-D-aspartate receptors by
dopamine D1 receptor ligands. Mol Pharmacol. 70:1761–1770. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Altar CA, Boyar WC and Kim HS:
Discriminatory roles for D1 and D2 dopamine receptor subtypes in
the in vivo control of neostriatal cyclic GMP. Eur J Pharmacol.
181:17–21. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Di Stefano A, Sozio P, Cacciatore I, et
al: Preparation and pharmacological characterization of
trans-2-amino-5(6)-fluoro-6(5)-hydroxy-1-phenyl-2,3-dihydro-1H-indenes
as D2-like dopamine receptor agonists. J Med Chem. 48:2646–2654.
2005.
|
|
29
|
Hoque KE, Indorkar RP, Sammut S and West
AR: Impact of dopamine-glutamate interactions on striatal neuronal
nitric oxide synthase activity. Psychopharmacology (Berl).
207:571–581. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morris RG, Hagan JJ and Rawlins JN:
Allocentric spatial learning by hippocampectomised rats: a further
test of the ‘spatial mapping’ and ‘working memory’ theories of
hippocampal function. Q J Exp Psychol B. 38:365–395.
1986.PubMed/NCBI
|
|
31
|
Park DJ and West AR: Regulation of
striatal nitric oxide synthesis by local dopamine and glutamate
interactions. J Neurochem. 111:1457–1465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sammut S, Dec A, Mitchell D, Linardakis J,
Ortiguela M and West AR: Phasic dopaminergic transmission increases
NO efflux in the rat dorsal striatum via a neuronal NOS and a
dopamine D(1/5) receptor-dependent mechanism.
Neuropsychopharmacology. 31:493–505. 2006. View Article : Google Scholar
|
|
33
|
Siuciak JA, McCarthy SA, Chapin DS, et al:
Genetic deletion of the striatum-enriched phosphodiesterase PDE10A:
evidence for altered striatal function. Neuropharmacology.
51:374–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Calabresi P, Centonze D, Gubellini P,
Marfia GA, Pisani A, Sancesario G and Bernardi G: Synaptic
transmission in the striatum: from plasticity to neurodegeneration.
Prog Neurobiol. 61:231–265. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Centonze D, Gubellini P, Pisani A,
Bernardi G and Calabresi P: Dopamine, acetylcholine and nitric
oxide systems interact to induce corticostriatal synaptic
plasticity. Rev Neurosci. 14:207–216. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kawaguchi Y: Neostriatal cell subtypes and
their functional roles. Neurosci Res. 27:1–8. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
West AR and Grace AA: Opposite influences
of endogenous dopamine D1 and D2 receptor activation on activity
states and electrophysiological properties of striatal neurons:
studies combining in vivo intracellular recordings and reverse
microdialysis. J Neurosci. 22:294–304. 2002.
|
|
38
|
Galati S, D’angelo V, Scarnati E, et al:
In vivo electrophysiology of dopamine-denervated striatum: focus on
the nitric oxide/cGMP signaling pathway. Synapse. 62:409–420. 2008.
View Article : Google Scholar : PubMed/NCBI
|














