Introduction
Atherosclerosis, a state of excessive oxidative
stress, is characterized by endothelial dysfunction.
Atherosclerotic plaques, which have a crucial role in the
occurrence and progression of atherosclerosis, have been reported
to preferentially settle at the branches, bifurcations or inner
curvatures of the artery, where shear stress is low (1,2).
Shear stress, a dragging force generated by blood flow, exerts a
variety of effects on endothelial function and contributes to the
focal location of atherosclerotic lesions (3). Several studies have identified that
the expression of endothelial nitric oxide synthase (eNOS) protein
is markedly increased in high shear stress regions and decreased by
low shear stress (LSS) (4–6). eNOS, a critical enzyme that converts
l-arginine to l-citrulline and nitric oxide (NO), is important in
maintaining endothelial function (7), as the release of NO affects the
formation, progression and heterogeneity of atherosclerotic plaques
(8,9). The regulation of eNOS activity
involves phosphorylation at multiple serine and threonine residues.
The phosphorylation of eNOS-Ser1177 near the carboxy-terminus is
required for eNOS activation, and eNOS-Ser633, located in the
flavin mononucleotide binding domain, also activates eNOS and is
pivotal for the maintenance of NO biosynthesis. eNOS activation is
inhibited by eNOS-Thr495, which interferes with the integration of
calmodulin at the eNOS calmodulin-binding domain (6,8,10).
Atherogenic events, including decreased NO
bioactivity and enhanced formation of reactive oxygen species
(ROS), are tightly associated with atherosclerosis formation. The
accumulation of ROS promotes apoptosis, leading to high endothelial
cell (EC) turnover, which promotes hotspots of increased
endothelial permeability and results in a preferred cellular
deposition to oxidized low density lipoproteins (11). Additionally, oxidative stress
contributes to vascular injury and variously pathological
processes, including atherosclerosis and thrombosis (12).
Resveratrol (RSV), a natural polyphenol in grapes,
pomegranates and peanuts, first attracted the attention of
investigators when it was correlated to the various biological
activities of red wine. RSV possesses diverse antiatherogenic
effects, including inhibiting low-density lipoprotein oxidation,
ameliorating cell apoptosis, disrupting platelet aggregation and
suppressing inflammatory factor secretion (13–15).
Furthermore, studies have reported that RSV has promising
neuroprotective and mitochondrial-improving functions, may
attenuate aging and prevent the onset of numerous chronic diseases
(16–18). In addition, RSV has the ability to
scavenge free oxygen radicals and has cytoprotective effects
(18–20). Several studies indicated that RSV
is able to enhance NO biosynthesis, which may be associated with
the extracellular signal-regulated kinase (ERK) signaling pathway
(21–23).
The regulatory mechanisms of RSV in the occurrence
of endothelial dysfunction induced by LSS have largely remained
elusive. In the present study, it was demonstrated that ERK/eNOS is
activated by LSS and that RSV inhibits LSS-induced oxidative stress
by suppressing ERK/eNOS signaling. Therefore, RSV, as a potential
regulator of eNOS, has a protective role in LSS-induced development
of atherosclerosis lesion.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs; Type
Culture Collection of the Chinese Academy of Sciences, Shanghai,
China) were cultured at 37°C in a humidified incubator with 5%
CO2 and maintained in RPMI-1640 culture medium
supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT,
USA). When grown to 90% confluence, cells were trypsinized,
harvested, resuspended and seeded onto a 0.1% gelatin-coated glass.
Following adherence, monolayer cells grown on the glass were used
for the flow experiments.
Shear stress study
A parallel-plate flow chamber (Shanghai Medical
Instrumentation College, Shanghai, China) that exerts a continuous
flow was established by sandwiching a silicon gasket between two
stainless steel plates with a cover slip sink in the base plate.
The chamber and all parts of the circuit tubes were sterilized
prior to the placement of the glass (50×30 mm) with monolayer cells
into the flow chamber. The parameter settings were as follows: Flow
chamber height, 0.56 mm and pump rate, 60 times/min. The value of
shear stress was adjusted by modulating the flow of after-loading
and was automatically calculated using the information from the
pressure transducers. The shear stress in our flow study was 2
dynes/cm2.
MTT assay
An MTT assay was employed to determine cell
viability. Following exposure to LSS for different durations or
treatment with different concentrations of RSV (Sigma, St. Louis,
MO, USA) for 1 h, cells were incubated in MTT (0.5 mg/ml; Sigma)
for 3 h. Following that, cells were washed twice with ice-cold PBS
and DMSO was added to solubilize the converted dye. Finally, DMSO
with formazan was collected and the absorbance was measured at 490
nm by a spectrophotometer (NanoDrop 2000; Thermo Scientific,
Waltham, MA, USA).
Detection of ROS and NO
MitoSOX™ is a live-cell permeant prober and
selectively detects superoxide in mitochondria. In the
mitochondria, MitoSOX is oxidized by superoxide and acquires red
fluorescent properties. 4,5-diaminofluorescein diacetate (DAF-2DA),
a membrane-permeable probe, enters cells and is converted into a
product with green fluorescence in the presence of NO. In the
present study, the glass with a monolayer of confluent cells was
placed into the flow chamber and the cells were subjected to LSS of
2 dynes/cm2 for 60 min with serum-free RPMI-1640 culture
medium as the perfusate. For the RSV group, cells were pretreated
with RSV for 30 min and subjected to a stress of 2
dynes/cm2 with serum-free RPMI-1640 medium containing
the same concentration of RSV as the cycle fluid. To measure ROS
and NO, cells following the flow study were gently washed twice
with ice-cold phosphate-buffered saline (PBS) and incubated with
either MitoSOX (5 μmol/l; Invitrogen Life Technologies, Carlsbad,
CA, USA) or DAF-2DA (5 μmol/l; Beyotime Institute of Biotechnology,
Jiangsu, China) at 37°C for 20 min under the exclusion of light.
Following this, the cells were washed twice with ice-cold PBS and
their nuclei were labeled with DAPI. The images were obtained using
a fluorescence microscope (Olympus IX71; Olympus, Tokyo,
Japan).
Measuring apoptotic cells
The terminal deoxynucleotidyl transferase dUTP nick
end labeling (TUNEL) reaction preferentially labels DNA strand
breaks which are produced during apoptosis. In the present study,
the cells exposed to LSS with RSV treated or not, were fixed in 4%
paraformaldehyde and permeabilized in 0.1% sodium citrate
containing 0.1% Triton X-100. Following blockade with 3%
H2O2 for 10 min, the cells were incubated
with TUNEL (Roche Diagnostics, Indianapolis, IN, USA) reaction
mixture at room temperature for 1.5 h. TUNEL-positive cells which
glowed green fluorescence were captured by a fluorescence
microscope.
Western blot analysis
Monolayer cells on a glass were exposed to LSS for
different durations with serum free RPMI-1640 medium as the cycle
fluid. In the inhibition group, cells were incubated with PD98059
(50 μmol/ml; Sigma), an inhibitor of ERK, prior to flow exposure.
Following the flow experiment, cells were lysed in a cocktail of
radioimmunoprecipitation assay buffer (50 mM Tris-HCl pH 7.5, 75 mM
NaCl, 15 mM EGTA, 1 mM dithiothreitol, 0.1% Tween-20, 60 mM
glycerophosphate, 1 mM NaF, 0.2 mM sodium orthovanadate and 2 mM
sodium pyrophosphate; Beyotime Institute of Biotechnology),
proteinase inhibitor (Sigma) and phosphatase inhibitor (Roche
Diagnostics), and lysates were clarified by centrifugation at
12,000 × g for 15 min at 4°C. Protein concentrations were
quantified by a bicinchoninic acid protein assay according to the
manufacturer’s instructions (KeyGen Biotech. Co. Ltd., Nanjing,
China). In total, 60 μg protein was separated by 10% SDS-PAGE and
transferred to a polyvinylidene fluoride membrane which was then
incubated overnight at 4°C with primary antibody. Following this,
the membrane was washed and incubated with horseradish
peroxidase-conjugated secondary antibody for 1 h at room
temperature. Following a second wash, the membranes were developed
using enhanced chemiluminescence substrates and the band
intensities were analyzed using Image J software (National
Institutes of Health, Bethesda, MD, USA). Goat anti-rabbit
IgG-horseradish peroxidase-coupled secondary antibody (1:2,000) and
primary rabbit antibodies against phospho-eNOS-Thr495 (1:1,000),
phospho-eNOS-Ser1177 (1:1,000), total eNOS (1:1,000), ERK 1/2
(1:1,000), phospho-ERK 1/2 (1:2,000) and GAPDH (1:2,000) were
purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Phospho-eNOS-Ser633 rabbit antibody (1:500) was obtained from Abcam
(Cambridge, MA, USA).
Determination of superoxide dismutase
(SOD) activity
The SOD assay is based on the formation of formazan
from the reaction of
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt and a superoxide radical produced intracellularly,
which is assayed at 450 nm. Cells exposed to LSS for 60 min with or
without ERK inhibition were lysed, scraped and centrifuged at
12,000 × g for 15 min at 4°C. The supernatant was collected and the
SOD activity was analyzed using a Total SOD assay kit with WST-1
according to the manufacturer’s instructions (Beyotime Institute of
Biotechnology).
Measurement of intracellular lactate
dehydrogenase (LDH)
LDH, an enzyme that leaks from membrane-disturbed
cells, was analyzed to confirm the protection of RSV on LSS-treated
cells. Cells subjected to LSS for 60 min with or without RSV
treatment were lysed, scraped and centrifuged. Subsequently, the
supernatant was collected and the concentration was determined.
Intracellular LDH was measured using an LDH assay kit according to
the manufacturer’s instructions (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China).
Quantitative polymerase chain reaction
(qPCR)
Total RNA was reverse transcribed into cDNA using a
PrimeScript® 1st strand cDNA Synthesis kit (Takara,
Dalian, China) and amplification was conducted by SYBR®
Premix Ex Taq™ (Takara). eNOS mRNA expression levels were analyzed
by qPCR (Applied Biosystems, Foster City, CA, USA) in three
independent experiments. The conditions were as follows: 38°C for 2
min and amplification at 95°C for 10 min, followed by 40 cycles at
95°C for 15 sec and annealed at 60°C for 1 min. The primers
targeting human eNOS were 5′-TGTTTCTGTCTGCATGG-3′ (forward) and
5′-TGGCTGGTAGCGGAAGG-3′ (reverse), and those targeting human GAPDH
were 5′-GACCTGACCTGCCGTCTA-3′ (forward) and
5′-AGGAGTGGGTGTCGCTGT-3′ (reverse) (Invitrogen Life Technologies).
The relative expression levels of genes were calculated according
to the 2−ΔΔCT method and eNOS mRNA was normalized to the
expression of GAPDH.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Student’s t-test was used to analyze data between two
groups and analysis of variance was utilized to compare data from
more than two groups. P<0.05 was considered to indicate a
statistically significant difference. Statistical analyses were
performed by SPSS, version 16.0 (SPSS, Inc. Chicago, IL, USA).
Results
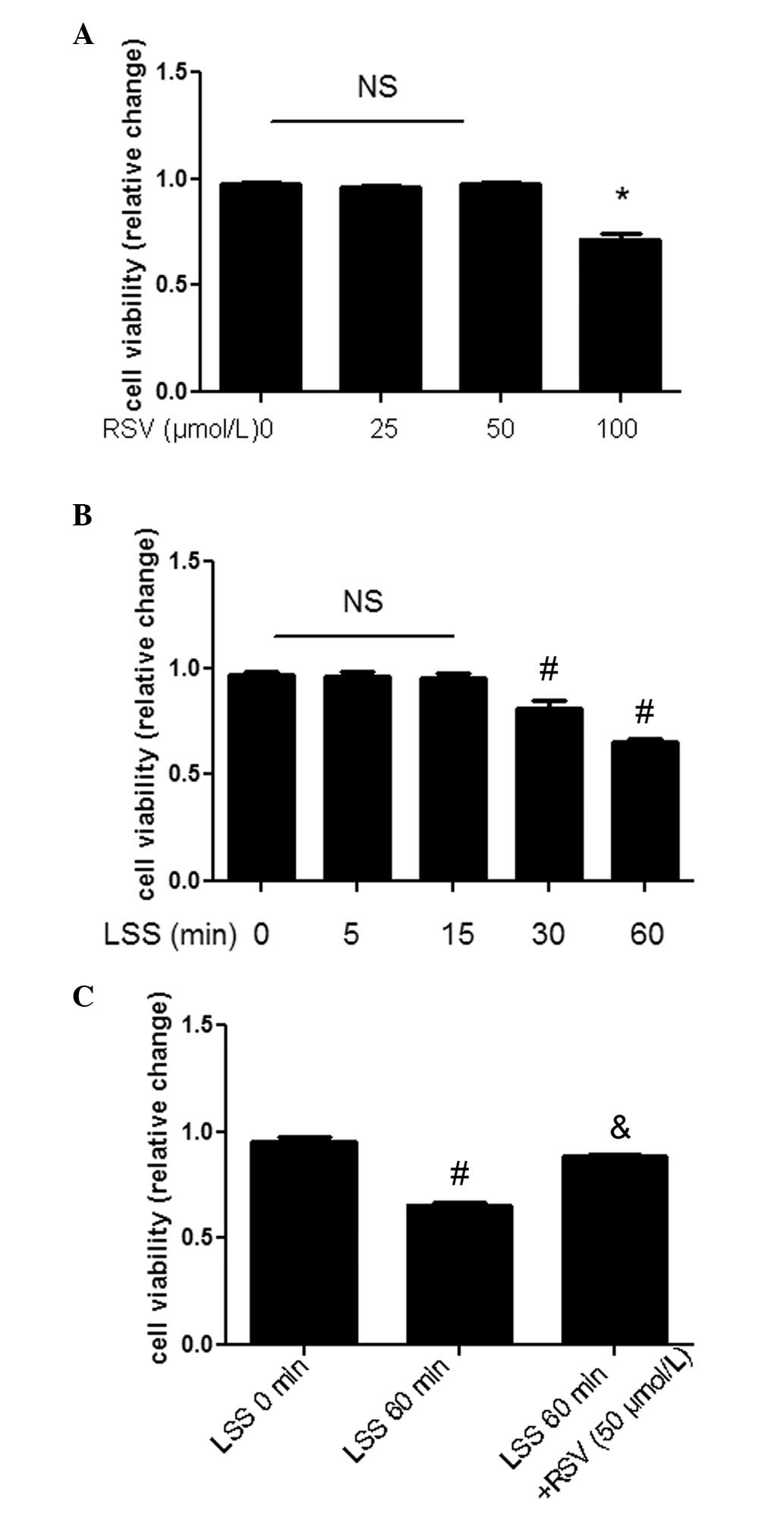
RSV restores LSS-mediated decreases in
cell viability
Cell viability was evaluated by an MTT assay. In
order to determine the proper concentration, RSV at concentrations
of 0–100 μmol/l was used. RSV at 100 μmol/l exhibited a cytotoxic
effect (Fig. 1A). Cell viability
was markedly decreased in the presence of LSS (Fig. 1B), which was restored by 50 μmol/l
RSV (Fig. 1C). Considering these
data, RSV at a concentration of 50 μmol/l was used in the present
study.
RSV attenuates LSS-induced oxidative
stress
First, the effects of LSS and RSV on ROS production
in ECs were investigated. Following 60 min of exposure to LSS, the
ROS levels exhibited a marked increase, which was attenuated by RSV
treatment. However, RSV did not affect the levels of ROS in the
static cultured cells (Fig. 2A).
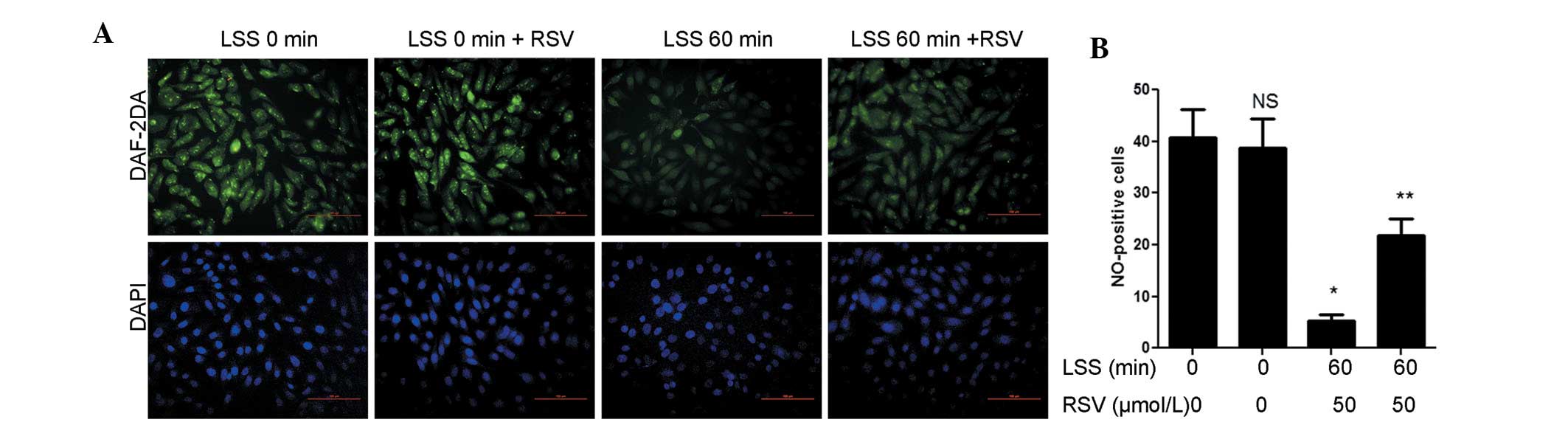
Due to the fact that excess ROS would impair eNOS function,
intracellular NO levels were measured with DAF-2DA. NO levels
decreased following 60 min of LSS compared with the LSS 0 min group
and were significantly increased following RSV treatment. Similar
to the ROS levels, the levels of NO were not affected by RSV under
static conditions (Fig. 3A).
Considering that oxidative stress-induced apoptosis indicates
endothelial dysfunction, TUNEL staining was used to detect
apoptotic cells. Cellular apoptosis was markedly increased by LSS,
which was attenuated with RSV treatment (Fig. 4A). These data suggested that RSV
exhibited antioxidant effects against LSS-induced oxidative
damage.
LSS activates eNOS-The495 and -Ser1177 in
a time-dependent manner
To determine whether LSS activates eNOS at Thr495,
Ser1177 and Ser633, which are closely associated with eNOS
activity, multi-phosphorylation sites of eNOS were examined after
the cells were subjected to LSS for different durations (0, 5, 15,
30 and 60 min). LSS significantly activated eNOS at Thr495 and
Ser1177 sites in a time-dependent manner, while it had no effect
eNOS-Ser633 (Fig. 5A). However,
acute nutritional deprivation may affect protein phosphorylation.
For instance, the substitution of culture medium supplemented with
10% FBS with serum-free medium may induce stress. To examine this
possibility, eNOS-Thr495 activation in cells starved for 30 min and
cells treated with LSS for 30 min were compared (Fig. 5C). As expected, the activation of
eNOS-Thr495 was due to the effect of LSS and was not a result of
serum-free stimulus.
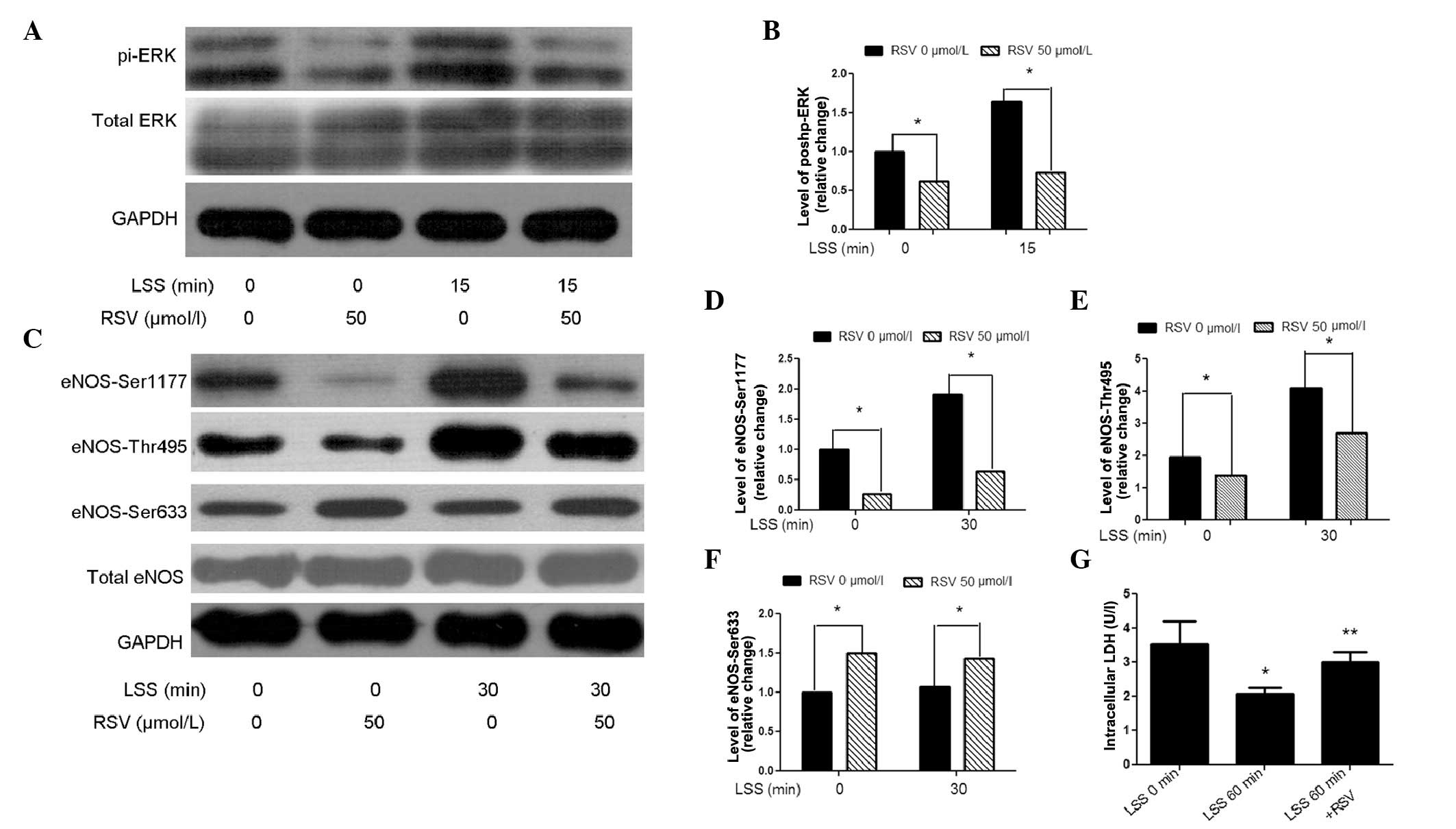
ERK inhibition suppresses LSS-mediated
phosphorylation of eNOS-Thr-495
With LSS, the activation of ERK reached a maximum
level at 15 min (Fig. 6A). To
investigate the hypothesis that ERK may serve as an upstream
modulator of LSS-mediated eNOS, PD98059, an inhibitor of ERK, was
used to treat the cells prior to the initiation of LSS for 30 min.
To ensure that an effective ERK inhibition was achieved,
phospho-ERK expression was analyzed, which was 78% reduced in cells
pre-incubated with PD98059 (50 μmol/l) compared with the control.
Subsequently, the effect of LSS on eNOS-Thr495 was examined and the
results demonstrated that ERK inhibition suppressed LSS-mediated
activation of eNOS-Thr495 (Fig.
6E). Additionally, SOD activity was measured to analyze the
antioxidant cellular defenses of ERK inhibition and the result
revealed that PD98059 induced an increase in SOD activity, as
compared with the control conditions (Fig. 6G). In conclusion, LSS-induced
oxidative stress is regulated by the ERK/eNOS-Thr495 signaling
pathway.
RSV blocks LSS-induced activation of
ERK/eNOS
The preliminary results suggested the presence of a
signaling pathway via LSS/ERK/eNOS. With the aim of investigating
the role of RSV, phospho-ERK expression in the presence or absence
of RSV in LSS-treated cells was evaluated. Compared with the
control, RSV decreased ERK activation at LSS 15 min (Fig. 7A). As the maximum LSS-mediated
stimulation of the phosphorylation of eNOS-Thr495 occurred at 30
min (Fig. 5A), this time-point was
selected to examine the effect of RSV on eNOS and a decrease in
eNOS-Thr495 levels was identified. In RSV-treated cells, eNOS
Ser-633 was increased, while it was not altered following a short
exposure to LSS. Unexpectedly, RSV deactivated LSS-phosphorylated
eNOS-Ser1177 (Fig. 7C). LSS has
been reported to increase cell permeability (26), which allows simple access for RSV
to the cell, thus resulting in an altered activation of eNOS.
Therefore, LDH, which was released from the permeability-increased
cells, was tested. The intracellular LDH assay demonstrated that
RSV restored LSS-mediated increases in cell permeability (Fig. 7G). These results suggested that the
antioxidant effect of RSV may partly be due to the suppression of
ERK/eNOS-Thr495.
Acute exposure to LSS does not alter the
expression of eNOS mRNA
qPCR was performed to investigate whether LSS
contributed to the change in eNOS mRNA expression. No significant
difference in eNOS mRNA levels was observed following LSS exposure
for 0 and 60 min in the presence or absence of RSV (Fig. 8). This result suggested that LSS
modulated eNOS in a post-transcriptional manner. Additionally,
activated protein phosphorylation independent from altered gene
expression is likely to have contributed to the modulation of eNOS
in the short time flow experiment.
Discussion
In the present study, the antioxidant effect of RSV
on LSS-treated cells was investigated and the possible mechanism
that may be involved in the modulation of eNOS was examined. The
results demonstrated that i) LSS evokes increased oxidative stress
in ECs which may be due to the activation of ERK/eNOS-Thr495 and
ii) RSV alleviates LSS-induced oxidative damage via suppressing the
ERK/eNOS-Thr495 signaling pathway.
Increases in ROS levels have been well-defined as a
mechanism for NO scavenging, leading to endothelial dysfunction
(24). Multiple factors have been
identified to enhance ROS formation in ECs, including mitochondrial
dysfunction and eNOS uncoupling (25). By specific detection of
mitochondrial superoxide by MitoSOX, the present study demonstrated
that LSS augmented ROS generation. Oxidative stress causes cellular
injury, and initiates apoptosis, which disrupts the integrity of
the endothelium, increases endothelial permeability, and therefore
leads to a preferred cellular deposition to lipids (26). Using TUNEL staining, apoptosis
caused by LSS was identified, which indicated that LSS caused
oxidative damage.
Three decades ago, Furchgott and Zawadzki (27) provided the first evidence that ECs
produced a factor which caused vascular smooth muscle relaxation.
Considering the observation was associated with NO, eNOS had since
been intensely studied (28).
Cheng et al (4) reported an
association between the distribution of shear stress and
differential activation of eNOS, which prompted us to investigate
the effects of LSS on eNOS multi-phosphorylation sites (4). Activation of Ser1177 or Ser633
promoted eNOS, while phosphorylation at Thr-495 had an inhibitory
effect (7). Based on the above
results and the evidence from the present study that LSS promoted
ROS formation and decreased NO production, the phosphorylation at
eNOS-Ser1177, Ser633 and Thr495 in ECs exposed to LSS was examined.
The results demonstrated that the activation of eNOS-Ser633
remained unchanged and that Thr495 was elevated at 5 min and peaked
at 30 min following the onset of flow. Unexpectedly, a short
exposure to LSS evoked eNOS-Ser1177, which contradicted the
original hypothesis that LSS inhibited eNOS-Ser1177 due to
promoting the release of NO (6,10).
Greif et al (29)
identified phosphatase protein phosphatase 2A (PP2A) as a key
determinant of eNOS dephosphorylation at Ser1177 and Thr495 sites.
Furthermore, an inhibitor of PP2A augmented overall levels of eNOS
phosphorylation and a mutation at Thr495 deactivated Ser1177. In
conclusion, it was proposed that PP2A is involved in LSS-activated
eNOS and that short-term LSS activates Ser1177 in a
Thr495-dependent manner; however, further study is required to
fully elucidate this.
MAPK signaling is important in a wide range of
cellular processes. ERK, one of the major factors in the MAPK
family, responds to a variety of extracellular stresses (30). First, the effect of LSS on ERK was
examined and it was observed that ERK was activated and peaked at
15 min. To identify the upstream regulator of eNOS, PD98059 (50
μmol/ml) was used to pre-incubate cells for 30 min prior to the
flow study. The results suggested that PD98059 suppressed
LSS-induced activation of eNOS-Thr495 and restored SOD activity.
These results corresponded with those obtained in other studies,
including that by Huang et al (30), who identified that ERK affects eNOS
under shear stress in a mouse model, and a study by Ford and Rush
(25) also reported that
activation of VEGF/ERK affects NO synthesis.
RSV is a natural polyphenol that has been
demonstrated to attenuate oxidative stress (17,20,31).
Substantial evidence suggested that the antioxidant effect of RSV
is attributed to the activation of sirtuins, which are mainly
located in the mitochondria (17).
Several other studies have demonstrated that RSV may increase NO
availability through the attenuation of ROS production (32). However, the mechanism by which RSV
affects eNOS in LSS-treated cells is elusive. The present findings
suggested that RSV attenuated oxidative stress and cell apoptosis,
and affected ERK/eNOS signaling, suggesting that RSV improved the
release of NO by blocking ERK/eNOS-Thr495. Furthermore, RSV was
identified to regulate MAPK in several other studies (30,33).
Skrobuk et al (34)
reported that an acute exposure to RSV inhibited AMPK activity,
which may explain why RSV deactivated eNOS-Ser1177 in the present
study (34). Additionally, the
manner of RSV-regulated eNOS deactivation was in accordance with
that mediated by PP2A (29). To
eliminate the possibility that the antioxidant effect of RSV may be
a result of increased cell permeability generated by LSS (11), intracellular LDH was measured. The
results revealed that RSV restored the integrity of the cell
membrane. Under static conditions, RSV affected the signaling of
ERK/eNOS. However, the ROS and NO levels, as well the apoptotic
rate of the cells, were independent of RSV under static conditions.
RSV failed to affect oxidative damage in static cultured cells,
suggesting that mechanisms other than eNOS may be associated with
the modulation of oxidative stress.
In conclusion, it is proposed that i) LSS causes
oxidative stress via ERK/eNOS-Thr495 and ii) RSV restores oxidative
damage through suppressing ERK/eNOS-Thr495. Furthermore, PP2A may
be a key regulator in LSS-affected eNOS and that LSS activates
Ser1177 in a Thr495-dependent way, which are hypotheses that
require further study.
There were certain limitations to the present study.
Atherosclerosis is considered to be a chronic and complex process,
and therefore, an extended exposure time in vivo should be
applied. The present study suggested that LSS-induced oxidative
damage was associated with ERK/eNOS-Thr495. PI3K/PKD/eNOS-Ser1177
and PKA/eNOS-Ser633 have been reported to be involved in long-term
high shear stress (35,36). High shear stress was also
identified to stimulate nuclear export of histone deacetylase 5 and
activate eNOS (37). However,
these signaling pathways are characterized by large and complicated
networks, and other key factors cannot be excluded, which requires
further investigation.
Acknowledgements
This study was generously supported by a grant from
the National Natural Science Foundation of China (no.
81270191).
References
|
1
|
Caro CG, Fitz-Gerald JM and Schroter RC:
Arterial wall shear and distribution of early atheroma in man.
Nature. 223:1159–1160. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
VanderLaan PA, Reardon CA and Getz GS:
Site specificity of atherosclerosis: site-selective responses to
atherosclerotic modulators. Arterioscler Thromb Vasc Biol.
24:12–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qin X, Tian J, Zhang P, Fan Y, Chen L,
Guan Y, Fu Y, Zhu Y, Chien S and Wang N: Laminar shear stress
up-regulates the expression of stearoyl-CoA desaturase-1 in
vascular endothelial cells. Cardiovasc Res. 74:506–514. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng C, Tempel D, Oostlander A, Helderman
F, Gijsen F, Wentzel J, van Haperen R, Haitsma DB, Serruys PW, van
der Steen AF, de Crom R and Krams R: Rapamycin modulates the eNOS
vs. shear stress relationship. Cardiovasc Res. 78:123–129. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng C, van Haperen R, de Waard M, van
Damme LC, Tempel D, Hanemaaijer L, van Cappellen GW, Bos J, Slager
CJ, Duncker DJ, van der Steen AF, de Crom R and Krams R: Shear
stress affects the intracellular distribution of eNOS: direct
demonstration by a novel in vivo technique. Blood. 106:3691–3698.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dimmeler S, Fleming I, Fisslthaler B,
Hermann C, Busse R and Zeiher AM: Activation of nitric oxide
synthase in endothelial cells by Akt-dependent phosphorylation.
Nature. 399:601–605. 1999. View
Article : Google Scholar
|
|
7
|
Sessa WC: eNOS at a glance. J Cell Sci.
117:2427–2429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Le Brocq M, Leslie SJ, Milliken P and
Megson IL: Endothelial dysfunction: from molecular mechanisms to
measurement, clinical implications, and therapeutic opportunities.
Antioxid Redox Signal. 10:1631–1674. 2008.PubMed/NCBI
|
|
9
|
Atkins GB and Simon DI: Interplay between
NF-kappaB and Kruppel-like factors in vascular inflammation and
atherosclerosis: location, location, location. J Am Heart Assoc.
2:e0002902013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mount PF, Kemp BE and Power DA: Regulation
of endothelial and myocardial NO synthesis by multi-site eNOS
phosphorylation. J Mol Cell Cardiol. 42:271–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cancel LM and Tarbell JM: The role of
mitosis in LDL transport through cultured endothelial cell
monolayers. Am J Physiol Heart Circ Physiol. 300:H769–H776. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu W, Ying H, Tong F, Zhang C, Quan Y and
Zhang Y: Protective effect of the silkworm protein 30Kc6 on human
vascular endothelial cells damaged by oxidized low density
lipoprotein (Ox-LDL). PLoS One. 8:e687462013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo H, Chen Y, Liao L and Wu W:
Resveratrol protects HUVECs from oxidized-LDL induced oxidative
damage by autophagy upregulation via the AMPK/SIRT1 pathway.
Cardiovasc Drugs Ther. 27:189–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malinowska J, Oleszek W, Stochmal A and
Olas B: The polyphenol-rich extracts from black chokeberry and
grape seeds impair changes in the platelet adhesion and aggregation
induced by a model of hyperhomocysteinemia. Eur J Nutr.
52:1049–1057. 2013. View Article : Google Scholar
|
|
15
|
Takizawa Y, Kosuge Y, Awaji H, Tamura E,
Takai A, Yanai T, Yamamoto R, Kokame K, Miyata T, Nakata R and
Inoue H: Up-regulation of endothelial nitric oxide synthase (eNOS),
silent mating type information regulation 2 homologue 1 (SIRT1) and
autophagy-related genes by repeated treatments with resveratrol in
human umbilical vein endothelial cells. Br J Nutr. 110:2150–2155.
2013. View Article : Google Scholar
|
|
16
|
Quincozes-Santos A, Bobermin LD, Latini A,
Wajner M, Souza DO, Goncalves CA and Gottfried C: Resveratrol
protects C6 astrocyte cell line against hydrogen peroxide-induced
oxidative stress through heme oxygenase 1. PLoS One. 8:e643722013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Price NL, Gomes AP, Ling AJ, Duarte FV,
Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro
JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel
R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA and Sinclair
DA: SIRT1 is required for AMPK activation and the beneficial
effects of resveratrol on mitochondrial function. Cell Metab.
15:675–690. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rajapakse AG, Yepuri G, Carvas JM, Stein
S, Matter CM, Scerri I, Ruffieux J, Montani JP, Ming XF and Yang Z:
Hyperactive S6K1 mediates oxidative stress and endothelial
dysfunction in aging: inhibition by resveratrol. PLoS One.
6:e192372011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kao CL, Chen LK, Chang YL, Yung MC, Hsu
CC, Chen YC, Lo WL, Chen SJ, Ku HH and Hwang SJ: Resveratrol
protects human endothelium from H(2)O(2)-induced oxidative stress
and senescence via SirT1 activation. J Atheroscler Thromb.
17:970–979. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arunachalam G, Yao H, Sundar IK, Caito S
and Rahman I: SIRT1 regulates oxidant- and cigarette smoke-induced
eNOS acetylation in endothelial cells: role of resveratrol. Biochem
Biophys Res Commun. 393:66–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Becatti M, Taddei N, Cecchi C, Nassi N,
Nassi PA and Fiorillo C: SIRT1 modulates MAPK pathways in
ischemic-reperfused cardiomyocytes. Cell Mol Life Sci.
69:2245–2260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Elíes J, Cuíñas A, García-Morales V,
Orallo F and Campos-Toimil M: Trans-resveratrol simultaneously
increases cytoplasmic Ca(2+) levels and nitric oxide release in
human endothelial cells. Mol Nutr Food Res. 8:1237–1248. 2011.
|
|
23
|
Min Z, Kang L, Lin L, Jinghua F, Junna S
and Baolin L: Resveratrol restores lysophosphatidylcholine-induced
loss of endothelium-dependent relaxation in rat aorta tissue
coinciding with inhibition of extracellular-signal-regulated
protein kinase activation. Phytother Res. 12:1762–1768. 2010.
View Article : Google Scholar
|
|
24
|
Qiu X, Brown K, Hirschey MD, Verdin E and
Chen D: Calorie restriction reduces oxidative stress by
SIRT3-mediated SOD2 activation. Cell Metab. 12:662–667. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ford RJ and Rush JW: Endothelium-dependent
vasorelaxation to the AMPK activator AICAR is enhanced in aorta
from hypertensive rats and is NO and EDCF dependent. Am J Physiol
Heart Circ Physiol. 300:H64–H75. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Conklin BS, Vito RP and Chen C: Effect of
low shear stress on permeability and occludin expression in porcine
artery endothelial cells. World J Surg. 31:733–743. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Furchgott RF and Zawadzki JV: The
obligatory role of endothelial cells in the relaxation of arterial
smooth muscle by acetylcholine. Nature. 288:373–376. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Palmer RM, Ferrige AG and Moncada S:
Nitric oxide release accounts for the biological activity of
endothelium-derived relaxing factor. Nature. 327:524–526. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Greif DM, Kou R and Michel T:
Site-specific dephosphorylation of endothelial nitric oxide
synthase by protein phosphatase 2A: evidence for crosstalk between
phosphorylation sites. Biochemistry. 41:15845–15853. 2002.
View Article : Google Scholar
|
|
30
|
Huang A, Yang YM, Yan C, Kaley G, Hintze
TH and Sun D: Altered MAPK signaling in progressive deterioration
of endothelial function in diabetic mice. Diabetes. 61:3181–3188.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carrizzo A, Puca A, Damato A, Marino M,
Franco E, Pompeo F, Traficante A, Civitillo F, Santini L, Trimarco
V and Vecchione C: Resveratrol improves vascular function in
patients with hypertension and dyslipidemia by modulating NO
metabolism. Hypertension. 62:359–366. 2013. View Article : Google Scholar
|
|
32
|
Schmitt CA, Heiss EH and Dirsch VM: Effect
of resveratrol on endothelial cell function: molecular mechanisms.
Biofactors. 36:342–349. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chan CM, Chang HH, Wang VC, Huang CL and
Hung CF: Inhibitory effects of resveratrol on PDGF-BB-induced
retinal pigment epithelial cell migration via PDGFRβ, PI3K/Akt and
MAPK pathways. PLoS One. 8:e568192013.PubMed/NCBI
|
|
34
|
Skrobuk P, von Kraemer S, Semenova MM,
Zitting A and Koistinen HA: Acute exposure to resveratrol inhibits
AMPK activity in human skeletal muscle cells. Diabetologia.
55:3051–3060. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Boo YC, Hwang J, Sykes M, Michell BJ, Kemp
BE, Lum H and Jo H: Shear stress stimulates phosphorylation of eNOS
at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol
Heart Circ Physiol. 283:H1819–H1828. 2002.PubMed/NCBI
|
|
36
|
Boo YC, Sorescu G, Boyd N, Shiojima I,
Walsh K, Du J and Jo H: Shear stress stimulates phosphorylation of
endothelial nitric-oxide synthase at Ser1179 by Akt-independent
mechanisms: role of protein kinase A. J Biol Chem. 277:3388–3396.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang W, Ha CH, Jhun BS, Wong C, Jain MK
and Jin ZG: Fluid shear stress stimulates phosphorylation-dependent
nuclear export of HDAC5 and mediates expression of KLF2 and eNOS.
Blood. 115:2971–2979. 2010. View Article : Google Scholar : PubMed/NCBI
|