Introduction
Glutamate is involved in fast excitatory
transmission and in neuronal functions, including plasticity and
cognitive processes, as well as in toxic events (1). The excitotoxic effects of glutamate
are largely mediated by increased Ca2+ influx through
activated N-methyl-d-aspartate (NMDA) receptors (2,3).
Initial neuronal death in the cerebral cortex may be due to an
excessive Ca2+ influx through NMDA receptors (4). A number of in vitro studies
indicated that at high concentrations, glutamate is a potent
neurotoxin capable of destroying neurons (3,5).
Diverse evidence has demonstrated that among
numerous signaling pathways involved in the survival and apoptosis
of neurons, three of the mitogen-activated protein kinase (MAPK)
signaling pathways, namely the p38 MAPK, c-Jun N-terminal kinase
and extracellular-regulated kinase (ERK) pathways, have been widely
studied (6,7). p38s are preferentially activated by
cell stress-induced signaling in response to factors including
oxidative stress, environmental stress and toxic chemical insults
(2). Therefore, p38 MAPKs are
considered ‘stress-activated protein kinases’ involved in cellular
signaling, inflammation, apoptosis, carcinogenesis and in the
pathogenesis of various diseases (3,5).
Additionally, a large number of neurotoxic chemicals have been
demonstrated to cause apoptosis in various neuronal cell
preparations mediated by the activation of p38 (6–8).
MAPK family members, including p38 MAPK, regulate diverse cellular
functions, including response to environmental stimuli and
apoptosis signaling (9). Studies
also indicated that the important functions of ERK1/2 and/or p38
MAPK in response to neurotoxic insults are mediated by
overstimulation of glutamate receptors in cell culture models
(7) and in cerebral ischemia
models (1).
Glucose/oxygen-deprivation has diverse consequences in cells,
including oxidative stress and excessive glutamate release reaching
toxic levels (4,10). However, the specific effects of
neurotoxic levels of glutamate on hippocampal slices have not been
characterized, particularly the mode of cell death and possible
signaling pathways involved in neurotoxicity (7–10).
The mechanisms underlying the mediation of
glutamate-induced neurotoxicity or excitotoxicity have yet to be
established; however, a substantial body of evidence suggests that
glutamate toxicity involves oxidative stress and apoptosis
(1,2,4,11).
Apoptosis is an important cell suicide program, which involves
caspase-dependent and -independent apoptotic pathways, and
dysregulation of apoptosis results in pathological conditions,
including cancer, autoimmune diseases and neurodegenerative
diseases (7). An increasing number
of studies imply that NMDA-induced apoptosis is triggered by
selective activation of NMDA receptors, and such a mechanism may be
mediated by calcium signaling pathways or the p38 pathway,
resulting in mitochondrial dysfunction and activation of caspase-3
(6,7,12).
Activation by cleavage of the proteolytic enzyme caspase is a key
step in the apoptotic program, and the upstream signaling pathways
leading to the assembly of protein death complexes activated by
caspases may or may not be dependent on mitochondria (1,10–12).
Therefore, the signaling pathways upstream of the disruption of the
mitochondrial membrane potential and caspase activation require
further study.
In the present study, the effects of the p38 MAPK
inhibitor SB203580 at different concentrations on the apoptosis of
NMDA-induced cerebral cortical neurons were observed to further
examine the possible mechanisms of NMDA-induced neuronal death.
Materials and methods
Cell isolation and culture
The cerebral cortical neurons were obtained from
brains of newborn Sprague-Dawley rats [Experimental Animal Center
of Liaoning Medical University, Jinzhou, China; Permission no. SCXK
(Liao) 2003–0007] within 24 h. Neonatal brain tissues were digested
with 2.5 g/l trypsin (Sigma, St. Louis, MO, USA) for 15 min at
37°C. Following centrifugation for 5 min at 126.87 × g, the tissues
were resuspended in high-glucose DMEM/F-12 (Hyclone, Logan, UT,
USA) containing 10% FBS and 10% heat-inactivated horse serum
(Hyclone), and triturated. The cell concentration was adjusted to
1×106/ml and the cells were seeded into plates coated
with poly-l-lysine (Sigma). The cells were incubated at 37°C in a
humidified incubator containing 5% CO2 for 24 h. The
medium was replaced and the cells were incubated in serum-free
neurobasal A medium (Gibco-BRL, Grand Island, NY, USA) supplemented
with 2% B27, 5 μmol/l glutamine, 100 U/ml penicillin and 100 U/ml
streptomycin (Guangzhou Weijia Technology Co., Ltd., Guangzhou,
Guangdong, China). On day two, the cultures were incubated with 2.5
mg/l cytosine arabinoside (Sigma-Aldrich) for 4 h to suppress the
growth of glial cells. The neurons grown for 7–8 days in
vitro (DIV) were used for further experimental observation. The
study was approved by the Animal Ethics Committee of Liaoning
Medical College (Jinzhou, China)
Grouping and intervention
Cortical neurons were cultured for 7 DIV prior to
drug treatment. The cultured cells were randomly assigned to five
groups: The control group, NMDA (50 μM) group and three groups
treated with NMDA (50 μM) in combination with three different
concentrations of the p38 MAPK inhibitor SB203580 (5, 10 and 20
μM). The different groups were prepared for the subsequent
experiments. The control group was incubated in an equal volume of
phosphate-buffered saline (PBS). Various concentrations of p38 MAPK
inhibitors were added 4 h prior to NMDA treatment. NMDA at 50 μM
(Sigma) was added to Mg2+-free Locke’s buffer in the
NMDA and SB203580 (Sigma-Aldrich) groups.
Analysis of cell viability
Cell viability was determined by an MTT assay. MTT
solution at 20 μl (Sigma-Aldrich), 5.0 g/l in PBS, was added to
each well of the 96-well plate (containing 100 μl of medium and
cells) 4 h prior to the end of incubation. The supernatant was
discarded and 150 μl dimethylsulfoxide was added to dissolve the
formazan, following which the culture plate was agitated. The
absorbance was measured at 570/630 nm using a microplate reader
(Sunrise™; Tecan, Grodig, Austria). The cell viability was
calculated from the optical density (OD) using the following
formula: treated group OD/control group OD × 100%.
Assessment of lactate dehydrogenase (LDH)
activity
LDH released from damaged cells into the cell
culture media was measured following treatment with NMDA and three
different concentrations of SB203580. A colorimetric assay was
used. According to this assay, the amount of formazan salt, which
was formed following conversion of lactate to pyruvate and then by
reduction of tetrazolium salt, was proportional to LDH activity in
the sample. Cell-free culture supernatants were collected from each
well and incubated with the mixture of the appropriate reagent
according to the manufacturer’s instructions (Cytotoxicity
Detection kit; Roche Diagnostics, Mannheim, Germany) for 20 min.
The intensity of the red color shown in the assay that was measured
at a wavelength of 490 nm using a multilabel counter system (Perkin
Elmer, Boston, MA, USA) was proportional to the LDH activity and
the number of damaged cells.
Apoptosis detection
Morphological evidence of apoptosis was obtained
using the acridine orange/ethidium bromide (AO/EB) staining method.
Primary cortical neuronal cells were cultured in six-well
flat-bottomed plates and allowed to adhere to the bottom of the
wells. The neurons grown for 7 DIV were treated with SB203580 for 4
h prior to exposure to NMDA. DMEM/F12 medium (10% FCS) was used as
a control for the cell lines. Following the indicated incubation
times, 25 μl AO/EB mixture (Sigma-Aldrich) was added to the cells
treated with or without NMDA. Then, the cells were examined using
fluorescence microscopy (Nikon Optiphot; Nikon, Tokyo, Japan) and
images were captured.
Immunohistochemical analysis
Cells were fixed with 4% paraformaldehyde and rinsed
with 0.1 M PBS. The cells were then incubated with primary
antibodies, which included phospho-p38 MAPK (p-p38MAPK; Cell
Signaling Technology, Danvers, MA, USA), B-cell lymphoma 2 (Bcl-2;
Abcam, Cambridge, MA, USA) and Bcl2-associated X (Bax; Millipore,
Billerica, MA, USA), in the blocking buffer for 48 h at 4°C. The
cultures were rinsed and incubated with the appropriate
biotinylated secondary antibody for 6 h and subsequently processed
with an avidin-biotin complex kit (Vectastain ABC kit; Vector
Laboratories, Burlingame, CA, USA) for another 1–2 h. Finally, the
reaction product was visualized with 0.05% 3,3′-diaminobenzidine as
the chromogen. Analysis was performed under a BX-60 microscope
(Olympus, Tokyo, Japan). The first antibodies were omitted in the
methodological control.
Western blot analysis
The total cell lysates (20 μg each) were separated
by 12.5% SDS-PAGE and transferred onto a polyvinylidene fluoride
(PVDF) membrane (Millipore) for 1.5 h at 400 mA using a Transphor
TE 62 (Hoefer, Inc., Holliston, MA, USA). The PVDF membranes were
then incubated with Bcl-2 (1:200), Bax (1:300), p-p38 MAPK (1:300)
and β-actin (1:2,000; Sigma-Aldrich) antibodies at 4°C for 2 h or
overnight. The membranes were washed three times in PBST (10 mM
NaH2PO4, 130 mM NaCl and 0.05% Tween 20) and
then probed with horseradish peroxidase-conjugated antibodies
(1:5,000; Sigma-Aldrich) for 1 h. The signals were visualized using
enhanced chemiluminesence western blot kits (Pierce Biotechnology,
Inc., Rockford, IL, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation
of three independent experiments. Comparisons among groups were
performed by one-way analysis of variance followed by Fisher’s
least significant difference, Student-Newman-Keuls or Dunnett’s T3
post-hoc multiple comparisons, when appropriate. SPSS 16.0 software
(SPSS Inc., Chicago, IL, USA) was used for the statistical
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
Neuron viability
Cultured cortical neurons were treated with or
without various concentrations of SB203580 for 4 h prior to
exposure to 50 μM NMDA. Apoptotic cells were assessed following 24
h using MTT assays. As illustrated in Fig. 1A, cell viability in the NMDA groups
and the SB203580-treated groups at three concentrations were 44±12,
49±10, 59±9 and 72±9%, respectively. The cell viability in the NMDA
groups was significantly lower than that in the control group
(P<0.01), while the cell viability in the groups treated with
the two highest concentrations of SB203580 was significantly
different from that in the NMDA groups (P<0.05, P<0.01). The
results demonstrated that the protective effect of SB203580 was
dose dependent, particularly in the high-dose group.
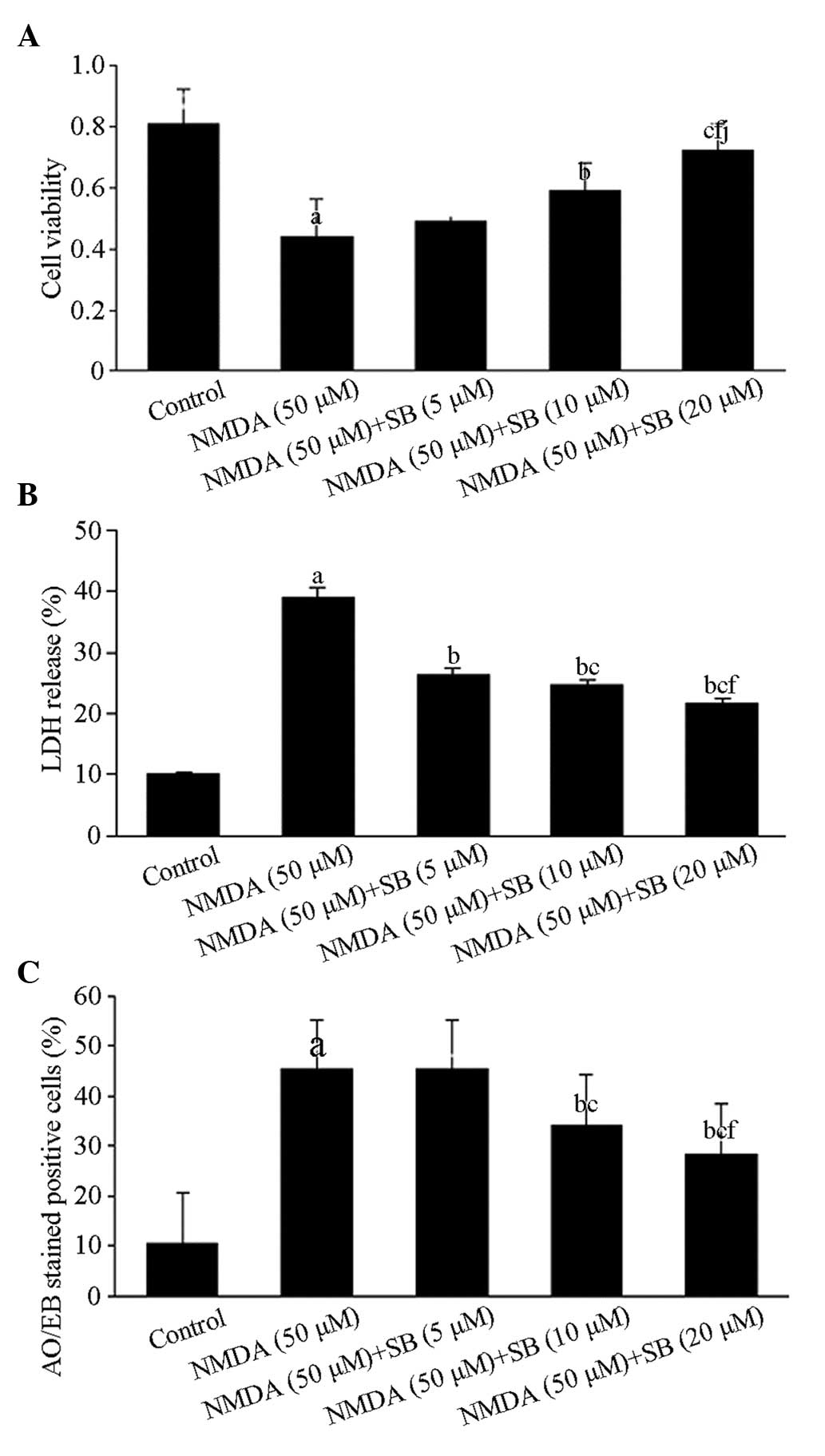 | Figure 1Effects of the p38 MAPK inhibitor
SB203580 on the viability of NMDA-induced neurons, LDH release and
neuronal apoptosis. (A) Cell viability following treatment with 50
μM NMDA. The values are expressed as the mean ± SD. n=6 for each
group. aP<0.01, vs the control group;
bP<0.05, cP<0.01, vs the NMDA injury
group; fP<0.01 vs the low dose group;
jP<0.05, vs the medium dose group. (B) LDH activity
24 h after cessation of NMDA-induced injury; the effect was dose-
and time-dependent. The values are expressed as the mean ± SD. n=6
for each group. aP<0.01, vs the control group;
bP<0.01, vs the NMDA injury group;
cP<0.01, vs the low dose group;
fP<0.01, vs the medium dose group. (C) Quantification
of apoptotic cells in rats from the control group, NMDA group and
SB203580 combined with NMDA groups. The values are expressed as the
mean ± SD. n=6 for each group. aP<0.01, compared with
the control group; bP<0.01, compared with the NMDA
group; cP<0.01, fP<0.01, comparisons
among SB203580 groups. MAPK, mitogen-activated protein kinase;
NMDA, N-methyl-d-aspartate; LDH, lactate dehydrogenase; SD,
standard deviation; AO/EB, acridine orange/ethidium bromide. |
LDH release
The exposure of cortical neurons to NMDA resulted in
LDH leakage. In the NMDA group, LDH levels increased to 39.10%
(P<0.01; Fig. 1B) compared with
that of the control group. The cells were pretreated with different
concentrations of SB203580 (5, 10 and 20 μmol/l) 4 h prior to NMDA
exposure. Following cells being incubated for 24 h, the LDH release
rates of the cells were reduced to 26.29, 24.59 and 21.65%,
respectively, which were significantly lower than the results of
the NMDA group (P<0.01; Fig.
1B).
Neuronal apoptosis
To investigate the type of cell death induced by
NMDA and observe the effects of SB202580 treatment, the cells were
stained with AO/EB, which allows the identification of viable,
apoptotic and necrotic cells based on color and appearance. As
shown in Figs. 1C and 2, the number of apoptotic neurons
increased significantly (P<0.01) compared with that in the
control groups. However, compared with the NMDA group, NMDA-induced
neuronal apoptosis significantly decreased (P<0.01) in the
SB203580 groups (5, 10 and 20 μm), particularly in the high-dose
group.
Immunohistochemical staining
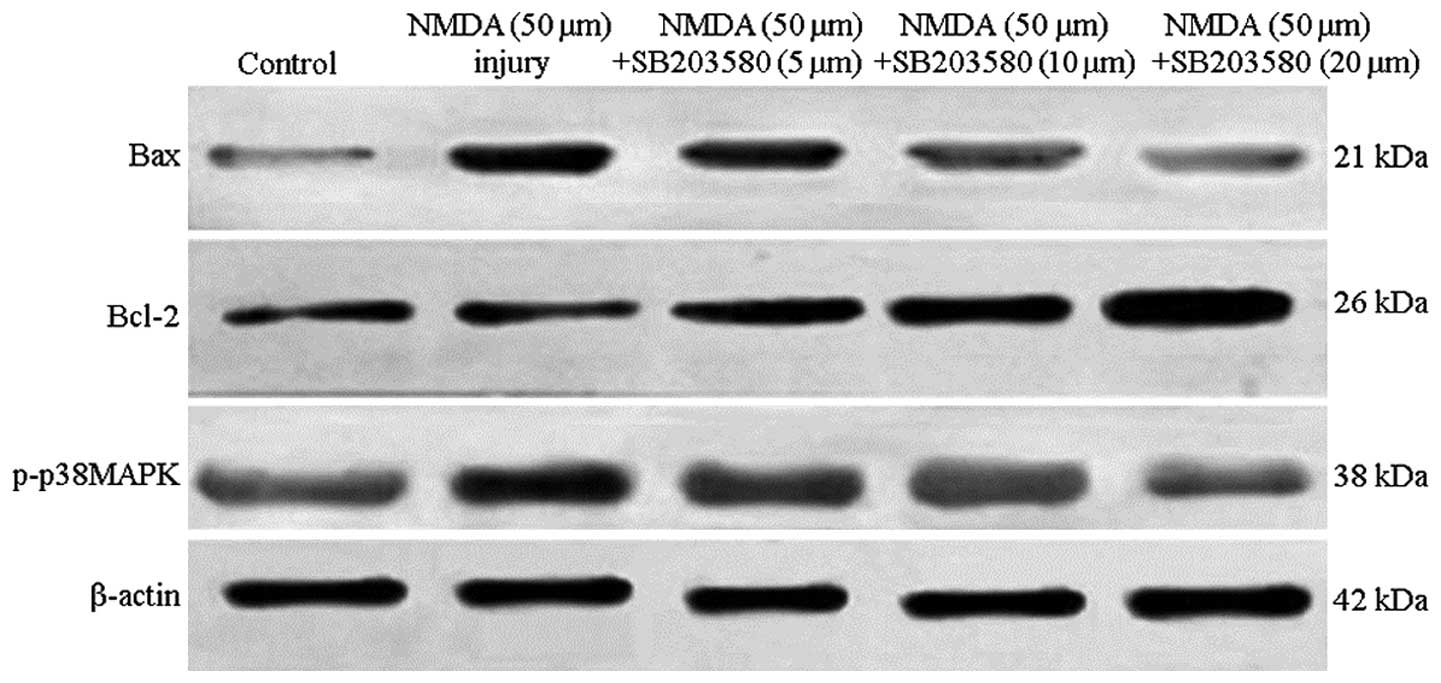
Relative changes in p38 MAPK, Bcl-2 and Bax were
determined by immunohistochemical staining, and the cytoplasm of
positive cells was stained brownish-yellow, as shown in Figs. 3 and 4A. NMDA treatment markedly increased the
expression of p-p38 MAPK and Bax and decreased the expression of
Bcl-2 compared with the control group. SB203580 treatment
significantly reduced the expression of p38 MAPK and Bax in
cortical neurons compared with the NMDA group (P<0.01). The
expression of Bcl-2 in cortical neurons was significantly increased
compared with that in the NMDA group (P<0.01), particularly in
the high-dose group. The results were dose-dependent. Compared with
the control group, the ratio of Bcl-2/Bax was significantly
decreased in the NMDA injury group (P<0.01). Compared with the
NMDA group, the Bcl-2/Bax ratio increased in the SB203580 groups at
different concentrations, which was statistically significant
(P<0.01).
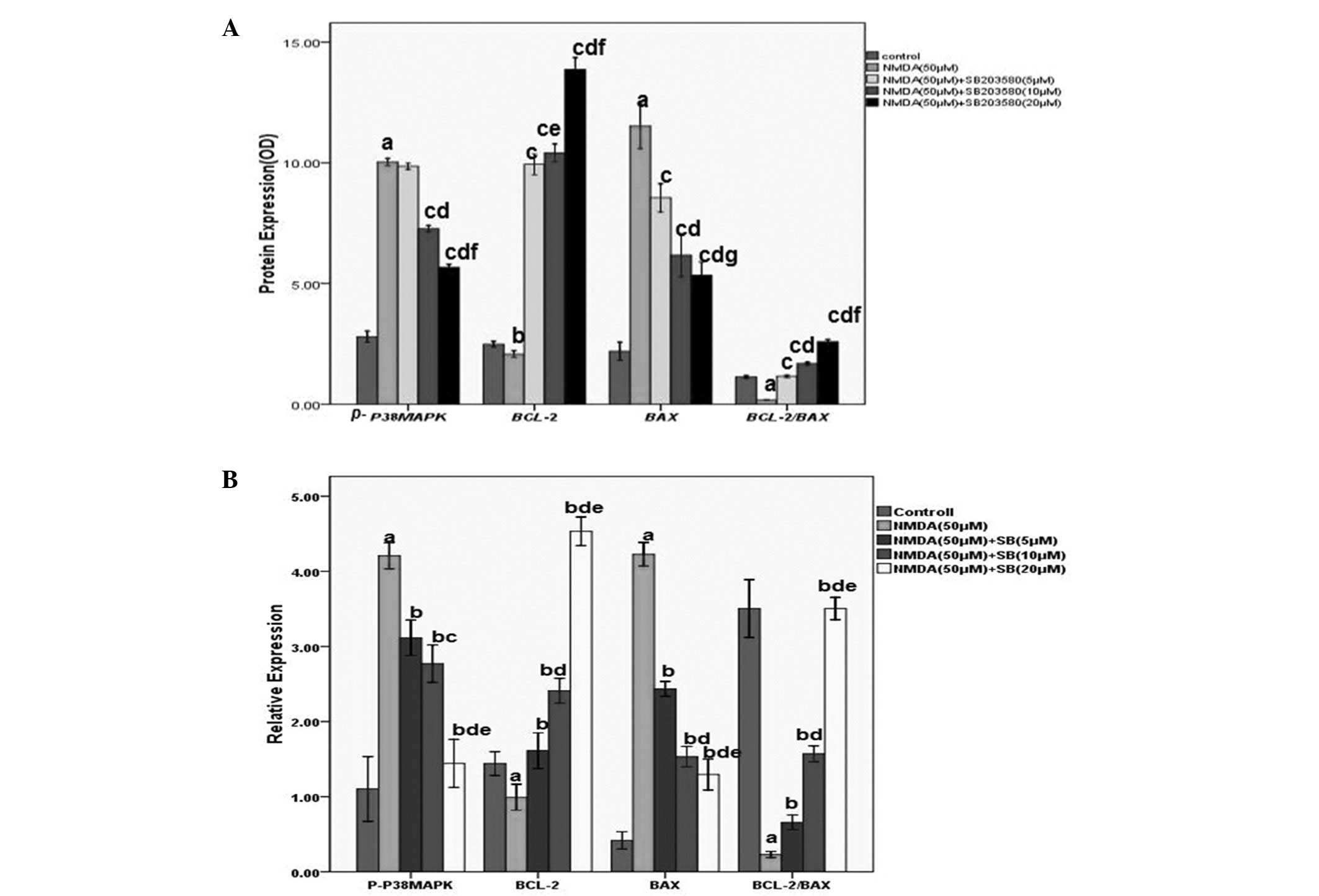 | Figure 4Quantitative analysis of relative
changes in p-p38 MAPK, Bcl-2 and Bax proteins by
immunohistochemical staining and western blot analysis. (A) Levels
of p-p38MAPK, Bcl-2, Bax and the ratio of the Bcl-2/Bax levels in
the cortical neurons with or without NMDA exposure or SB203580
immunization. The number in each column indicates the absorbance
value of the detected proteins. Values (means ± SD) are expressed
as the relative expression levels. aP<0.01,
bP<0.05 vs the control group; cP<0.01,
vs the NMDA injury group; fP<0.01,
jP<0.05 vs the low dose group; kP<0.05,
lP<0.05 vs the medium dose group. (B) Quantitative
analysis of relative changes in p-p38 MAPK, Bcl-2 and Bax proteins.
The values are expressed as the mean ± SD. n=6 for each group.
aP<0.01, vs the control group; bP<0.01
vs the NMDA injury group; cP<0.05,
fP<0.01, vs the low dose group;
kP<0.01, vs the medium dose group. MAPK,
mitogen-activated protein kinase; NMDA,
N-methyl-d-aspartate; OD, optical density; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X; SD, standard deviation. |
Western blot analysis
The levels of p-p38MAPK, Bcl-2, Bax and Bcl-2/Bax
were examined by western blot analysis. As shown in Figs. 4B and 5, NMDA significantly increased p-p38MAPK
and Bax levels as compared with the control group (P<0.01).
Bcl-2 levels decreased significantly compared with those in the
control group (P<0.01). p-p38MAPK and Bax protein levels were
markedly reduced and Bcl-2 was increased compared with the NMDA
group (P<0.01). Comparisons among different interventional
groups revealed significant differences (P<0.05, P<0.01) and
the protein expression levels were dose-dependent. Compared with
the control group, the ratio of Bcl-2/Bax significantly decreased
in the NMDA injury group (P<0.01). Compared with the NMDA group,
the ratio of Bcl-2/Bax was increased at different concentrations of
SB203580, with a significant difference (P<0.01).
Discussion
Among numerous signaling pathways involved in the
survival and apoptosis of neurons, MAPKs are a family of signaling
molecules involved in the transduction of extracellular stimuli
into intracellular responses in a wide variety of circumstances
(11). p38 MAPK is important in
inflammation and apoptosis and it is part of a signaling cascade
that has been implicated in neuronal death associated with kainic
acid (KA)-induced seizures (6). An
increasing amount of evidence suggests that excitotoxicity may also
have an important pathogenic function in processes that culminate
in programmed cell death. Glutamate-induced neurotoxicity is mainly
studied in cell cultures, in which glutamate evokes necrosis and/or
apoptosis depending on experimental conditions and, at low
concentrations, glutamate may induce apoptosis, however, not
necrosis (8,11,12).
However, the contribution of p38 MAPK to NMDA-induced apoptosis has
not been fully elucidated.
To evaluate the involvement of signaling pathways
dependent on p38 MAPK on the NMDA-induced damage to cortical
neurons, the selective inhibitor SB203580 was used. In the present
study, LDH and MTT reduction assays were used to quantitatively
estimate the effects of neuronal injury following NMDA exposure and
SB203580 treatment. As shown in Figs.
1 and 2, cell viability
significantly increased and LDH leakage markedly decreased compared
with the NMDA groups. The activation of p38 MAPK under different
circumstances, including degeneration induced by cerebral ischemia
(5), excitotoxicity induced by
glutamate in cerebellar granule neurons (3,13)
and KA in the hippocampus (6–8) and
methyl-mercury-induced neurotoxicity in cultured Neuro-2a cells
(9), demonstrates the direct
contribution of the p38 MAPK pathway to neuronal cell death. The
activation of p38 MAPK may be a key step in the induction of
cortical neuronal damage in response to the insult by NMDA, and
SB203580 demonstrated a dose-dependent protective effect from
NMDA-induced neuronal injury. Therefore, the present study
confirmed that the activation of p38 MAPK is involved in
NMDA-induced neuronal injury.
According to previous studies,
Ca2+-mediated activation of p38 MAPK causes excitotoxic
neuronal death (9,14). SB203580 was revealed to have
protective effects against NMDA-induced neuronal injury; however,
its optimal protective condition is not known. The detailed
molecular mechanisms by which NMDA induces p38 MAPK activation
leading to neuronal apoptosis remain controversial and elusive.
Thus, numerous mechanisms require further investigation, including
those of apoptotic proteins.
Apoptotic proteins, which include anti-apoptotic
Bcl-2 and pro-apoptotic Bax, may result either in the inhibition or
promotion of cell death (7,13).
Bcl-2 mitigates calcium entry and mitochondrial calcium overload
through regulation of l-type calcium channels, and these effects
may contribute to the maintenance of cell viability following
injury (15,16). Pro-apoptotic Bax promotes
cytochrome c release followed by the activation of caspases to
induce apoptotic cell death. The protein levels of the cell death
repressor Bcl-2 and cell death promoter Bax determine the threshold
for neuronal cell death (4,11,16).
Their expression is dynamically modulated at the onset of
neurodegeneration (17).
Furthermore, Bcl-2 expression and the activation of the p38 MAPK
pathway are involved in neuronal apoptosis induced under neurotoxic
conditions (5,18). The overexpression of Bcl-2 was able
to attenuate the cytotoxicity of NMDA-induced apoptosis, and was
able to extend cell survival time and maintain cell stability
(2,12,19).
A fine balance exists between apoptosis and
anti-apoptosis and this balance may be disrupted when cells are
affected by external chemical stimuli, and the apoptotic process is
able to be activated. The p38 MAPK signaling system for the
regulation of apoptosis, whether or not there is such a focal
point, requires further investigation. Studies have suggested that
the mechanism of estrogen-mediated neuroprotection involves the
regulation of mitochondrial Ca2+ and Bcl-2 expression
(20,15). Exposure of glutamate has also been
associated with increased cytosolic Ca2+ in cortical
neurons (15,21) and upregulation of pro-apoptotic
protein Bax in neuronal cell lines (1,22,23).
In the present study, immunohistochemical staining and western blot
analyses were used to assess the expression of p-p38MAPK, Bcl-2 and
Bax following NMDA treatment alone or in combination with SB203580.
As shown in Figs. 4 and 5, increased levels of p-p38MAPK and Bax
were observed following NMDA treatment, and the levels of Bcl-2
were downregulated. However, SB203580 was able to reverse this
effect, particularly at high doses. Together with the results of
the AO/EB staining, it was further verified that NMDA-induced
apoptosis is mediated in part by p-p38 MAPK activation. Numerous
in vitro studies have demonstrated that selective p38 MAPK
inhibitors protect hippocampal and cortical neuron cultures from
NMDA excitotoxicity (6,24), as well as cerebellar granular cell
culture from glutamate-induced apoptosis (1,11,25).
However, the findings of the present study agree with previous
results, supporting the hypothesis that the activation of p38 MAPK
may be a key step in the induction of hippocampal cell damage in
response to insults by NMDA.
In conclusion, the present study provided evidence
that cell death induced by NMDA in primary cortical neurons partly
proceeded via apoptosis. p38 MAPK served as an external apoptosis
triggering signal in neurons following NMDA treatment. Notably,
inhibition of p38 MAPK by SB203580 was able to prevent neuronal
cell apoptosis caused by NMDA. The results of the present study may
contribute to a better understanding of the mechanisms involved in
brain injury induced by NMDA. The results may also facilitate the
design of an improved strategy for developing biomedical therapies
for apoptosis control, as well as the control of the development of
disorders that involve p-p38 MAPK, Bcl-2 and Bax, including
ischemic cerebrovascular disease, epilepsy and others.
Acknowledgements
This study was supported by Liaoning Social
Development Key Projects (no. 2012225019) and Natural Science
Foundation (no. 2014022030) of the Scientific and Technological
Department of Liaoning Province. The authors would like to thank
the staff of the Experimental Teaching Center of Pharmacology and
Physiology, Liaoning Medical College and Analytical and Testing
Center of Liaoning Medical College, for their technical guidance
and support during experimentation.
References
|
1
|
Molz S, Decker H, Dal-Cim T, Cremonez C,
Cordova FM, Leal RB and Tasca CI: Glutamate-induced toxicity in
hippocampal slices involves apoptotic features and p38 MAPK
signaling. Neurochem Res. 33:27–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen J, Errico SL and Freed WJ: Reactive
oxygen species and p38 phosphorylation regulate the protective
effect of Δ9-tetrahydrocannabinol in the apoptotic
response to NMDA. Neurosci Lett. 389:99–103. 2005.
|
|
3
|
Yang XR, Sun P, Qin HP, Si PP, Sun XF and
Zhang C: Involvement of MAPK pathways in NMDA-induced apoptosis of
rat cortical neurons. Sheng Li Xue Bao. 64:609–616. 2012.PubMed/NCBI
|
|
4
|
Lu TH, Hsieh SY, Yen CC, et al:
Involvement of oxidative stress-mediated ERK1/2 and p38 activation
regulated mitochondria-dependent apoptotic signals in
methylmercury-induced neuronal cell injury. Toxicol Lett.
204:71–80. 2011. View Article : Google Scholar
|
|
5
|
Zhang R, Sun L, Hayashi Y, Liu X, Koyama
S, Wu Z and Nakanishi H: Acute p38-mediated inhibition of
NMDA-induced outward currents in hippocampal CA1 neurons by
interleukin-1beta. Neurobiol Dis. 38:68–77. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dunleavy M, Provenzano G, Henshall DC and
Bozzi Y: Kainic acid-induced seizures modulate Akt (SER473)
phosphorylation in the hippocampus of dopamine D2 receptor knockout
mice. J Mol Neurosci. 49:202–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Giordano G, Klintworth HM, Kavanagh TJ and
Costa LG: Apoptosis induced by domoic acid in mouse cerebellar
granule neurons involves activation of p38 and JNK MAP kinases.
Neurochem Int. 52:1100–1105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neoh CA, Wang RY, Din ZH, et al: Induction
of apoptosis by sinulariolide from soft coral through
mitochondrial-related and p38MAPK pathways on human bladder
carcinoma cells. Mar Drugs. 10:2893–2911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grethe S, Ares MP, Andersson T and
Pörn-Ares MI: p38 MAPK mediates TNF-induced apoptosis in
endothelial cells via phosphorylation and downregulation of
Bcl-x(L). Exp Cell Res. 298:632–642. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang HL, Dhar SS and Wong-Riley MT: p38
mitogen-activated protein kinase and calcium channels mediate
signaling in depolarization-induced activation of peroxisome
proliferator-activated receptor gamma coactivator-1alpha neurons. J
Neurosci Res. 88:640–649. 2010.
|
|
12
|
Yan Y, Bian JC, Zhong LX, Zhang Y, Sun Y
and Liu ZP: Oxidative stress and apoptotic changes of rat cerebral
cortical neurons exposed to cadmium in vitro. Biomed Environ Sci.
25:172–181. 2012.PubMed/NCBI
|
|
13
|
Sanchez A, Tripathy D, Yin X, Luo J,
Martinez J and Grammas P: Pigment epithelium-derived factor (PEDF)
protects cortical neurons in vitro from oxidant injury by
activation of extracellular signal-regulated kinase (ERK) 1/2 and
induction of Bcl-2. Neurosci Res. 72:1–8. 2012. View Article : Google Scholar
|
|
14
|
Guo RB, Wang GF, Zhao AP, Gu J, Sun XL and
Hu G: Paeoniflorin protects against ischemia-induced brain damages
in rats via inhibiting MAPKs/NF-kB-mediated inflammatory responses.
PLoS One. 7:e497012012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Śmiałowska M, Gołembiowska K, Kajta M,
Zieba B, Dziubina A and Domin H: Selective mGluR1 antagonist EMQMCM
inhibits the kainate-induced excitotoxicity in primary neuronal
cultures and in the rat hippocampus. Neurotox Res. 21:379–392.
2012.PubMed/NCBI
|
|
16
|
Tiwari M, Lopez-Cruzan M, Morgan WW and
Herman B: Loss of caspase-2-dependent apoptosis induces autophagy
after mitochondrial oxidative stress in primary cultures of young
adult cortical neurons. J Biol Chem. 286:8493–8506. 2011.
View Article : Google Scholar
|
|
17
|
Liu B, Zhang H, Xu C, et al:
Neuroprotective effects of icariin on corticosterone-induced
apoptosis in primary cultured rat hippocampal neurons. Brain Res.
1375:59–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu Z, Wang BR, Wang X, Kuang F, Duan XL,
Jiao XY and Ju G: ERK 1/2 and p38 mitogen-activated protein kinase
mediate iNOS-induced spinal neuron degeneration after acute
traumatic spinal cord injury. Life Sci. 79:1895–1905. 2006.
View Article : Google Scholar
|
|
19
|
Jiang W, Van Cleemput J, Sheerin AH, et
al: Involvement of extracellular regulated kinase and p38 kinase in
hippocampal seizure tolerance. J Neurosci Res. 81:581–588. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Segura Torres JE, Chaparro-Huerta V,
Rivera Cervantres MC, Montes-González R, Flores Soto ME and
Beas-Zárate C: Neuronal cell death due to glutamate excitotocity is
mediated by P38 activation in the rat cerebral cortex. Neurosci
Lett. 403:233–238. 2006.PubMed/NCBI
|
|
21
|
Miloso M, Scuteri A, Foudah D and Tredici
G: MAPKs as mediators of cell fate determination: an approach to
neurodegenerative diseases. Curr Med Chem. 15:538–548. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen S, Xu Y, Xu B, et al: CaMKII is
involved in cadmium activation of MAPK and mTOR pathways leading to
neuronal cell death. J Neurochem. 119:1108–1118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y and Bhavnani BR: Glutamate-induced
apoptosis in primary cortical neurons is inhibited by equine
estrogens via down-regulation of caspase-3 and prevention of
mitochondrial cytochrome c release. BMC Neurosci. 6:132005.
View Article : Google Scholar
|
|
24
|
Liu XW, Ma C, Xing RX, et al: The
calmodulin-dependent protein kinase II inhibitor KN-93 protects rat
cerebral cortical neurons from N-methyl-D-aspartic acid-induced
injury. NRR. 8:111–120. 2013.PubMed/NCBI
|
|
25
|
Xu B, Xu Z, Deng Y, Liu W, Yang H and Wei
YG: MK-801 protects against intracellular Ca(2+)
overloading and improves N-methyl-D-aspartate receptor expression
in cerebral cortex of methylmercury-poisoned rats. J Mol Neurosci.
49:162–171. 2013.PubMed/NCBI
|