Introduction
The proliferation and migration of vascular smooth
muscle cells (VSMCs), which is associated with neointimal
formation, is a critical pathological process in the formation of
vascular lesions and may result in atherosclerosis and restenosis
(1,2). Growth factors and cytokines produced
by several types of cells, including macrophages, endothelial cells
and VSMCs, contribute to the development and progression of
vascular lesions (3).
Platelet-derived growth factor (PDGF), a significant regulator of
mitogenesis, influences the proliferation of VSMCs through the
activation of extracellular signal-regulated kinase (ERK) 1/2,
c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase
(MAPK) and Akt signaling pathways (4,5).
VSMCs proliferate and migrate into the intimal layer
following vascular injury in atherosclerotic lesions (1). The proliferation of VSMCs is
primarily controlled by regulation of the cell cycle, which is
composed of four distinct sequential phases, known as
G0/G1, S, G2 and M (6). Following proliferative stimulation,
VSMCs enter S phase from the quiescent G0/G1
phase (6). This G1- to
S-phase transition is regulated by cyclin/cyclin-dependent kinase
(CDK) complexes, including cyclin D1/CDK4 and cyclin E/CDK2, which
are inhibited by the negative regulators p21WAF1 and p27KIP1
(7,8).
Matrix metalloproteinases (MMPs), including the
gelatinases MMP-2 and MMP-9, degrade type IV collagen, which leads
to the migration and invasion of VSMCs, resulting in the intimal
thickening that is characteristic of vascular plaque instability
(9). MMP expression is tightly
regulated at the transcriptional level by growth factors,
cytokines, hormones and tumor promoters (1,9).
In vitro and in vivo studies have shown that MMP-9
expression is critical in the progression of arterial lesions
(9–13). In VSMCs, MMP-9 expression is
regulated by various stimuli, including growth factors and
cytokines secreted by platelets, macrophages and VSMCs (9–15).
The thorns of Gleditsia sinensis are used in
traditional medicinal treatments and have been shown to exert
anti-cancer, anti-mutagenic, anti-allergenic, anti-microbial,
anti-human immunodeficiency virus and anti-inflammatory effects
(16–21). The primary components of
Gleditsia sinensis are stigmasterol (16), ellagic acid glycosides (22), flavonoids (17) and lupane acid (18). We have previously demonstrated that
the ethanol extract of Gleditsia sinensis thorns (EEGS) is
capable of inhibiting tumor necrosis factor α-induced VSMC
proliferation (23). However, the
molecular mechanism underlying the inhibitory effect of EEGS in the
proliferation and migration of PDGF-induced VSMCs is yet to be
elucidated. Therefore, the present study aimed to investigate the
inhibitory effects of EEGS on cell proliferation and migration in
PDGF-induced VSMCs.
Materials and methods
Materials
Polyclonal antibodies against cyclin E, CDK2 and
CDK4 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz,
CA, USA). Polyclonal antibodies against cyclin D1, p21WAF1, p53,
p27, ERK, phosphorylated (phospho)-ERK, p38 MAPK, phospho-p38 MAPK,
JNK, phospho-JNK, Akt, phospho-Akt and GAPDH were obtained from New
England Biolabs Inc. (Ipswich, MA, USA). The polyclonal MMP-9
antibody was obtained from Chemicon (Temecula, CA, USA). All
experimental procedures were and protocols were approved by the
Ethics Committee of Chung-Ang University (Anseong, Republic of
Korea).
Preparation of extract
A total of 100 g air-dried Gleditsia sinensis
thorns were crushed, prior to the addition of ethanol. Extraction
was performed by heating to 100°C. The extract was then
concentrated using a rotary evaporator, and lyophilized. The final
extract, which weighed 10 g (a collection rate of 10%), was diluted
in saline solution.
Cell cultures
Aortic smooth muscle cells were obtained from the
aortas of young male Sprague Dawley rat (8 weeks old, 200–250 g)
using enzymatic digestion, as described previously (23). VSMCs were isolated from the Sprague
Dawley rats. Briefly, the aortas were removed under sterile
conditions. Following being rinsed several times in Hanks’ balanced
salt solution, the adventitia was removed from the aortas and the
aortas were homogenized and digested using 5 ml digestion solution
(0.125 mg/ml elastase, 0.25 mg/ml soybean trypsin inhibitor, 10
mg/ml collagenase I, 2.0 mg/ml crystallized bovine albumin and 15
mM HEPES) at 37°C for 45 min. The cellular digests were filtered
through a sterile 100-μm nylon mesh, centrifuged at 184 × g for 10
min and washed twice in Dulbecco’s modified Eagle’s medium (DMEM)
containing 10% fetal calf serum, prior to being cultured in the
same medium. Experiments shown are representative of the results
from three independent cultures from each group of rats. VSMC
characterization was performed by immunofluorescence staining using
a monoclonal antibody against smooth muscle-α-actin (Sigma Aldrich,
St. Louis, MO, USA). These explants were incubated in DMEM
containing 10% fetal bovine serum (FBS), 2 mM glutamine, 50 μg/ml
gentamycin and 50 μl/ml amphotericin-B at 37°C in a humidified 5%
CO2 atmosphere. Cells were passaged every 3–5 days and
experiments were performed on cells at primary culture passages
five to eight. In the majority of experiments, cells at a
confluence of 80–90% were made quiescent by 24 h of incubation in
DMEM without FBS.
Cell viability assay
Growth-arrested VSMCs were incubated on 24-well
plates with EEGS for varying time-periods in the presence of PDGF.
Cell viability was determined using a modified MTT assay, which was
based on the conversion of tetrazolium salt
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxym-thoxyphenyl)-2-(4-sulfophenyl)-2-tetrazolium
to the formazan product by mitochondrial dehydrogenases (23). Formazan was quantified by measuring
the absorbance at 490 nm.
[3H]thymidine
incorporation
VSMCs, grown to near-confluence in 24-well tissue
culture plates, were made quiescent and treated with EEGS in the
presence of PDGF, as indicated. The [3H]thymidine
incorporation experiment was performed as described previously
(23).
Cell-cycle analysis using
fluorescence-activated cell sorting (FACS)
Cells were harvested, fixed in 70% ethanol and
stored at −20°C. Cells were then washed twice with ice-cold
phosphate-buffered saline (PBS) and incubated with RNase, the DNA
intercalating dye and propidium iodide. Cell-cycle phase analysis
was performed using a Becton Dickinson FACStar™ flow cytometer
equipped with Becton Dickinson Cell Fit software (BD Biosciences,
Franklin Lakes, NJ, USA).
Western blot analysis
Quiescence was induced in VSMCs, which were grown to
near-confluence in 100-mm tissue culture plates, prior to treatment
with EEGS in the presence of PDGF for varying durations at 37°C.
Cells were then washed twice with cold PBS and freeze-thawed in 250
μl lysis buffer [50 mmol/l HEPES (pH 7.5), 150 mmol/l NaCl, 1
mmol/l EDTA, 2.5 mmol/l ethylene glycol tetraacetic acid (EGTA), 1
mmol/l dithiothreitol (DTT), 10 mmol/l β-glycerophosphate, 1 mmol/l
NaF, 0.1 mmol/l Na3VO4, 0.1 mmol/l
phenylmethylsulfonyl fluoride (PMSF), 10% glycerol, 0.1% Tween-20,
10 μg/ml leupeptin and 2 μg/ml aprotinin]. Cell lysates were then
harvested into 1.5-ml tubes and placed on ice for 15 min, prior to
centrifugation at 10,786 × g for 20 min at 4°C. The protein
concentration of the supernatant was determined using the Bradford
Protein Assay (Bio-Rad, Hercules, CA, USA). Equal quantities of
cellular proteins were resolved by electrophoresis on a 0.1%
SDS-10% polyacrylamide gel under denaturing conditions. The
proteins were then electrophoretically transferred to
nitrocellulose membranes (Amersham Biosciences Corp., Piscataway,
NJ, USA). Following blocking in 10 mmol/l Tris-HCl (pH 8.0), 150
mmol/l NaCl and 5% (w/v) non-fat dry milk, membranes were incubated
with primary antibodies for 90 min and then further incubated with
peroxidase-conjugated secondary antibodies for 45 min.
Immunocomplexes were detected using a chemiluminescence reagent kit
(Amersham Biosciences Corp.). The experiments were repeated at
least three times (13).
Immunoprecipitation and immune-complex
kinase assays
Cell lysates were prepared using ice-cold lysis
buffer [50 mmol/l HEPES (pH 6.0), 150 mmol/l NaCl, 1 mmol/l EDTA,
2.5 mmol/l EGTA, 1 mmol/l DTT, 10 mmol/l β-glycerophosphate, 1
mmol/l NaF, 0.1 mmol/l Na3VO4, 0.1 mmol/l
PMSF, 10% glycerol, 0.1% Tween-20, 10 μg/ml leupeptin and 2 μg/ml
aprotinin] and sonicated twice for 10 sec using a Micro Ultrasonic
Cell Disrupter (Kontes; Kimble Chase, LLC, Vineland, NJ, USA) at
30% power and 4°C. Lysates were clarified by centrifugation at
10,000 × g for 5 min, and the supernatants were precipitated by
treatment with protein A-Sepharose beads precoated with saturating
quantities of the indicated antibodies at 4°C for 2 h. When
monoclonal antibodies were used, protein A-Sepharose was pretreated
with rabbit anti-mouse immunoglobulin G (Jackson ImmunoResearch
Laboratories, Inc., West Grove, MA, USA). Proteins that were
immunoprecipitated on the beads were washed four times with 1 ml
lysis buffer and twice with kinase buffer (50 mmol/l HEPES, 10
mmol/l MgCl2, 1 mmol/l DTT, 10 mmol/l
β-glycerophosphate, 1 mmol/l NaF and 0.1 mmol/l sodium
orthovanadate). The final pellet was resuspended in 25 μl kinase
buffer containing either 1 μg glutathione S-transferase
(GST)-retinoblastoma protein (pRb) C-terminal (pRb amino acids 769
to 921) fusion protein (Santa Cruz Biotechnology, Inc.) or 5 μg
histone H1 (Invitrogen Life Technologies, Carlsbad, CA,
USA), 20 μmol/l ATP and 5 μCi [γ32P]ATP (4,500 μCi/mmol;
ICN Pharmaceuticals Inc., Costa Mesa, CA, USA), and incubated for
20 min at 30°C with occasional mixing. The reaction was terminated
by the addition of 25 μl 2X Laemmli sample buffer and separated on
10 or 12.5% SDS-polyacrylamide gels. The migration of histone
H1 and GST-pRb was determined by Coomassie blue
staining, and phosphorylated pRb and histone H1 were
visualized.
Wound-healing migration assay
The growth-arrested cells were damaged using a
2-mm-wide tip. Cells were then treated with EEGS either alone or
with PDGF. Cells were subsequently allowed to migrate and images
were captured using an inverted microscope (magnification,
×40).
Invasion assay
The growth-arrested cells were resuspended with EEGS
either alone or with PDGF, in 100 μl medium. Cells were placed in
the upper section of the Transwell plate and incubated for 24 h.
Cells had to pass through a polycarbonate membrane that had a thin
layer of extracellular matrix (ECM)-like material with 8-μm pores.
The ability of the cells to invade the ECM-like material was
determined using a commercial cell invasion assay kit (Chemicon),
as described previously.
Zymography
The conditioned medium was electrophoresed in a
polyacrylamide gel containing 1 mg/ml gelatin. The gel was then
washed at room temperature for 2 h with 2.5% Triton X-100 and
maintained at 37°C overnight in a buffer containing 10 mM
CaCl2, 150 mM NaCl and 50 mM Tris-HCl (pH 7.5). The gel
was stained with 0.2% Coomassie blue and images were obtained using
a light box. Proteolysis was detected as a white zone in a dark
blue field.
Nuclear extracts and electrophoretic
mobility shift assay (EMSA)
Nuclear extracts were prepared as described
previously (13). Cultured cells
were collected by centrifugation and washed and suspended in a
buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1
mM EGTA, 1 mM DTT and 0.5 mM PMSF. After 15 min on ice, cells were
vortexed in the presence of 0.5% Nonidet P-40. The nuclear pellet
was then collected by centrifugation and extracted in a buffer
containing 20 mM HEPES (pH 7.9), 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA,
1 mM DTT and 1 mM PMSF for 15 min at 4°C.
The nuclear extract (10–20 μg) was preincubated at
4°C for 30 min with a 100-fold excess of an unlabeled
oligonucleotide spanning the −79 MMP-9 cis element of
interest. The sequences were as follows: AP-1,
CTGACCCCTGAGTCAGCACTT; Sp-1, GCCCATTCCTTCCGCCCCCAGATGAAGCAG and
NF-κB, CAGTGGAATTCCCCAGCC. The reaction was then incubated at 4°C
for 20 min in a buffer containing 25 mM HEPES (pH 7.9), 0.5 mM
EDTA, 0.5 mM DTT, 0.05 M NaCl and 2.5% glycerol, with 2 μg
poly(deoxyinosinic-deoxycytidylic) acid and 5 fmol
(2×104 cpm) Klenow end-labeled (32P-ATP)
30-mer oligonucleotide, which spanned the DNA binding site in the
MMP-9 promoter. The reaction mixture was electrophoresed at 4°C on
a 6% polyacrylamide gel using a Tris-borate-EDTA (89 mM Tris, 89 mM
boric acid and 1 mM EDTA) running buffer. The gel was rinsed with
water, dried and exposed to X-ray film overnight.
Statistical analysis
Where appropriate, data are expressed as the mean ±
standard error of the mean. Data were analyzed using factorial
analysis of variance and a Fisher’s least significant difference
test, where appropriate. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
EEGS inhibits PDGF-stimulated VSMC
proliferation
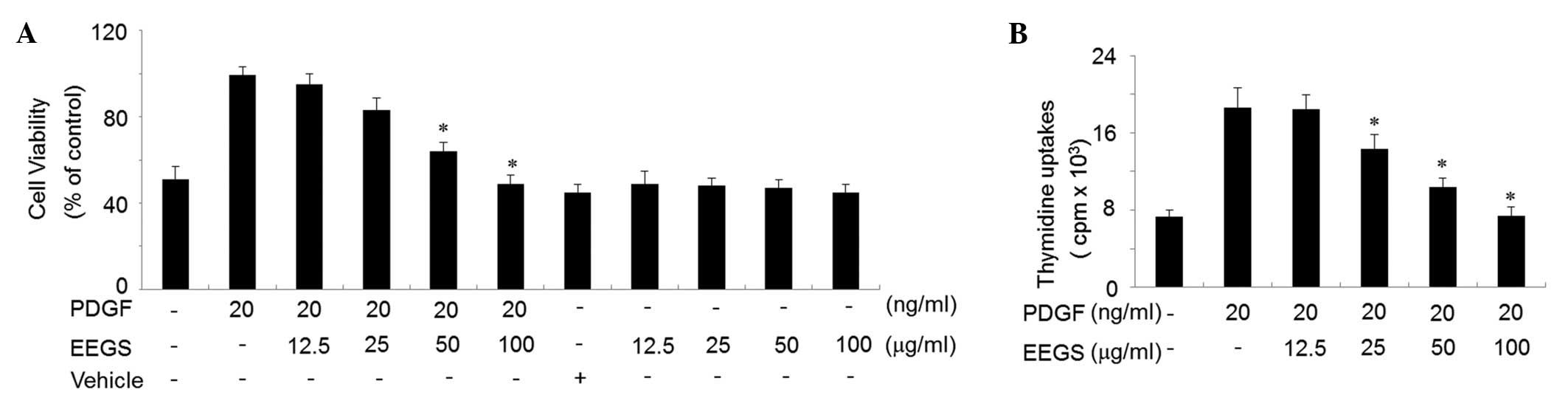
An MTT assay was used to assess the effect of EEGS
on cell viability. PDGF treatment for 24 h was observed to increase
VSMC viability approximately two-fold compared with the non-treated
control VSMCs (Fig. 1A). However,
treatment with EEGS (100 μg/ml) was found to inhibit the increase
in cell viability induced by PDGF to a controlled level (Fig. 1A). EEGS treatment either alone
(12.5–100 μg/ml) or with a vehicle (ethanol) was not observed to
affect cell viability (Fig. 1A).
To examine whether EEGS treatment inhibited PDGF-induced cell
proliferation, a thymidine uptake incorporation assay was used.
VSMCs were pretreated with 12.5, 25, 50 or 100 μg/ml EEGS for 40
min, then stimulated with PDGF (20 ng/ml) for 24 h. PDGF-treated
cells showed a significant increase in thymidine uptake compared
with the control cells (P<0.05; Fig. 1B). This PDGF-induced cell
proliferation was significantly inhibited by EEGS in a
concentration-dependent manner (P<0.05; Fig. 1B).
EEGS induces G1-phase
cell-cycle arrest in PDGF-stimulated VSMC proliferation
Flow cytometric analysis was used to assess whether
the anti-proliferative effect of EEGS was due to cell-cycle arrest
in a specific phase. PDGF treatment was observed to significantly
increase the proportion of VSMCs in the S and G2/M
phases of the cell cycle, with a concomitant decrease in the
proportion in G1 phase, compared with the control cells
(P<0.05; Table I). However,
EEGS (100 μg/ml) treatment in the presence of PDGF was observed to
markedly reduce the percentage of cells in the S and
G2/M phases, resulting in a significant accumulation of
cells in G1 phase, compared with the PDGF-treated VSMCs
(P<0.05; Table I). These data
indicate that EEGS treatment had an inhibitory effect on
PDGF-induced VSMC proliferation, through the suppression of
G1- to S-phase transition.
 | Table IFlow cytometric analysis of quiescent
vascular smooth muscle cells treated with PDGF and various
concentrations of EEGS. |
Table I
Flow cytometric analysis of quiescent
vascular smooth muscle cells treated with PDGF and various
concentrations of EEGS.
| Group |
G0/G1 phase (%) | S phase (%) | G2/M
phase (%) |
|---|
| Control | 75.51 | 16.52 | 7.97 |
| PDGF (20
ng/ml) | 53.24 | 30.79 | 15.97 |
| PDGF + EEGS (25
μg/ml) | 56.86 | 28.64 | 14.50 |
| PDGF + EEGS (50
μg/ml) | 68.68 | 17.52 | 13.80 |
| PDGF + EEGS (100
μg/ml) | 66.74 | 18.52 | 14.74 |
EEGS-induced G1-phase
cell-cycle arrest is associated with a decrease in cyclin-related
kinase activity
To investigate the mechanism of EEGS-induced
G1-phase cell-cycle arrest, the effects of EEGS on
cyclins and CDKs were examined. As shown in Fig. 2A, PDGF treatment was observed to
increase the expression of cyclin D1, cyclin E, CDK2 and CDK4 in
VSMCs, and these effects were significantly inhibited with the
addition of EEGS. The kinase activities of CDKs control cell-cycle
transition (6–8). Therefore, the kinase activities
associated with CDK2 and CDK4 were examined in EEGS-treated cells
in the presence of PDGF. Treatment of VSMCs with PDGF was observed
to significantly increase the kinase activities of the CDK2- and
CDK4-immunoprecipitates (Fig. 2B).
Furthermore, the PDGF-induced CDK2 and CDK4 activities were
inhibited by EEGS treatment (Fig.
2B).
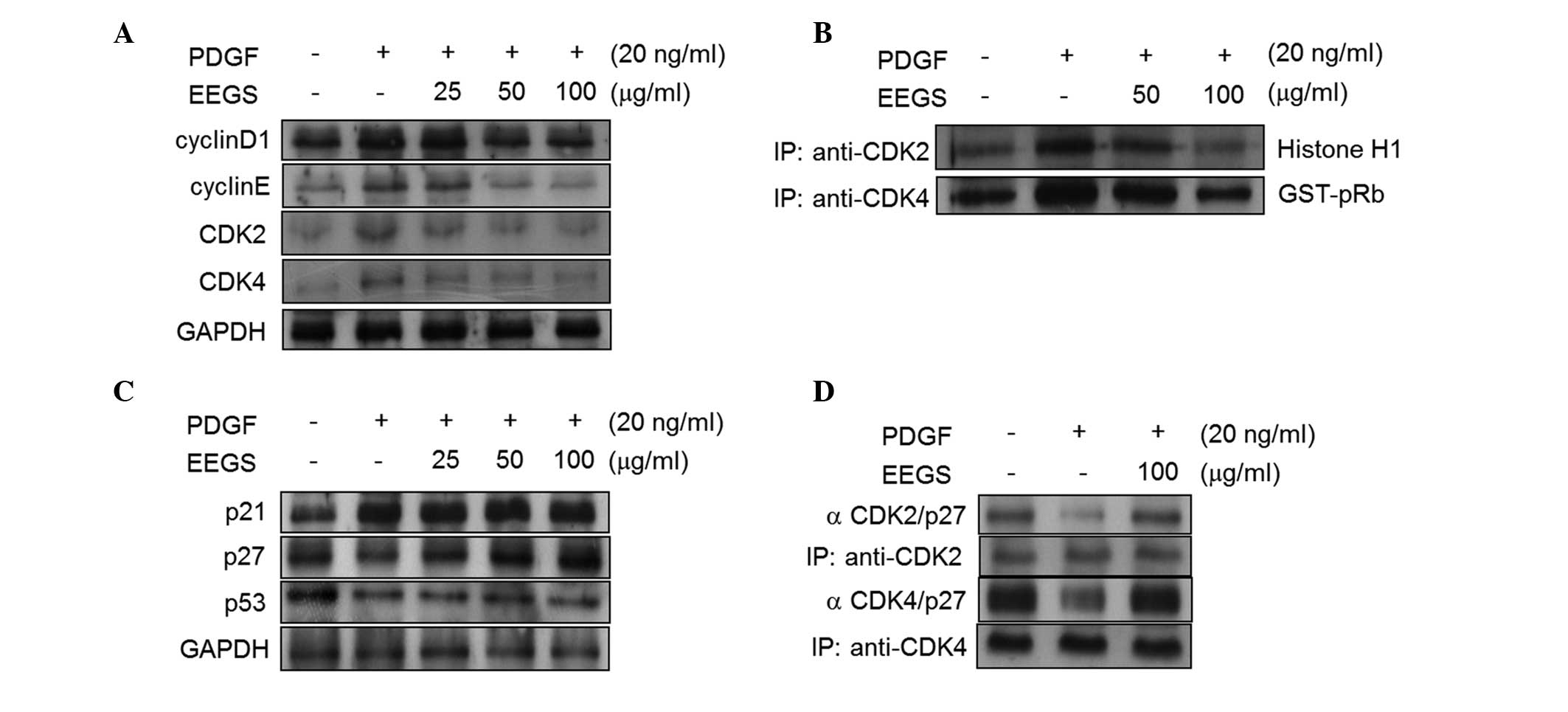 | Figure 2EEGS induces G1-phase cell
cycle arrest through the expression of p27KIP1 in PDGF-treated
VSMCs. Quiescent VSMCs were stimulated with PDGF (20 ng/ml) in the
presence or absence of the indicated concentrations of EEGS for 24
h. (A and C) Western blot analysis was performed using antibodies
specific for cyclin D1, cyclin E, CDK2, CDK4, p21WAF1, p27KIP1 and
p53. The results were normalized to GAPDH expression. (B) Total
cell lysates were immunoprecipitated using anti-CDK2 and -CDK4
antibodies. The kinase reaction was performed using histone
H1 (for CDK2) or GST-Rb (for CDK4) as substrates. (D)
Equal quantities of cell lysates were subjected to
immunoprecipitation using anti-CDK2 and -CDK4 antibodies, followed
by SDS-PAGE. Following electrophoresis, samples were transferred to
nitrocellulose membranes, and western blot analysis was performed
using an anti-p27KIP1 antibody. PDGF, platelet-derived growth
factor; EEGS, ethanol extract of Gleditsia sinensis thorns;
VSMC, vascular smooth muscle cell; CDK, cyclin-dependent kinase;
GST, glutathione S-transferase; pRb, retinoblastoma protein;
IP, immunoprecipitation. |
p27KIP1 expression is associated with
EEGS-induced G1-phase cell-cycle arrest
G1- to S-phase cell-cycle progression is
negatively regulated by cyclin-dependent kinase inhibitors (CDKIs),
including p27KIP1 (7,8). Therefore, the effect of EEGS on
p27KIP1 expression was assessed in PDGF-treated VSMCs. In the
serum-starved quiescent VSMCs, p27KIP1 was constitutively
expressed, and PDGF was observed to suppress its expression
(Fig. 2C). Pretreatment with EEGS
was found to reverse the PDGF-induced downregulation of p27KIP1
(Fig. 2C). By contrast, p21WAF1
expression increased with the addition of PDGF and remained
unchanged with the addition of EEGS (Fig. 2C). Furthermore, PDGF reduced the
protein expression of p53, and this was not affected by EEGS
addition (Fig. 2C). It is well
established that a reduction in kinase activity is involved in the
increased interaction between p27KIP1 and CDKs (7,8).
Therefore, the effects of EEGS on p27KIP1-CDK interactions were
assessed using an immunoprecipitation assay. The interaction
between CDK2 and p27KIP1 was observed to be downregulated in
PDGF-treated VSMCs (Fig. 2D). This
interaction was upregulated in the presence of EEGS (Fig. 2D). Under similar experimental
conditions, levels of the p27KIP1-CDK4 complexes were also
increased in EEGS-treated cells following PDGF stimulation
(Fig. 2D). These results indicate
that p27KIP1 may be involved in EEGS-induced G1-phase
cell-cycle arrest in PDGF-treated VSMCs.
EEGS inhibits PDGF-stimulated Akt
phosphorylation in VSMCs
The effect of PDGF treatment on the phosphorylation
of MAPK and Akt in VSMCs was next investigated. After 10 min PDGF
treatment, an increase in the phosphorylation of ERK1/2, JNK,
p38MAPK and Akt was observed, with no effect on the total protein
levels of these molecules (Fig.
3). The inhibitory effect of EEGS on PDGF-induced ERK1/2, JNK,
p38MAPK and Akt phosphorylation was also investigated. Pretreatment
of VSMCs with EEGS was found to significantly inhibit Akt
phosphorylation in PDGF-treated VSMCs (Fig. 3). However, PDGF-induced
phosphorylation of ERK1/2, JNK and p38MAPK was not affected by the
addition of EEGS (Fig. 3). These
results demonstrate that EEGS may inhibit PDGF-induced VSMC
proliferation through the inhibition of Akt phosphorylation.
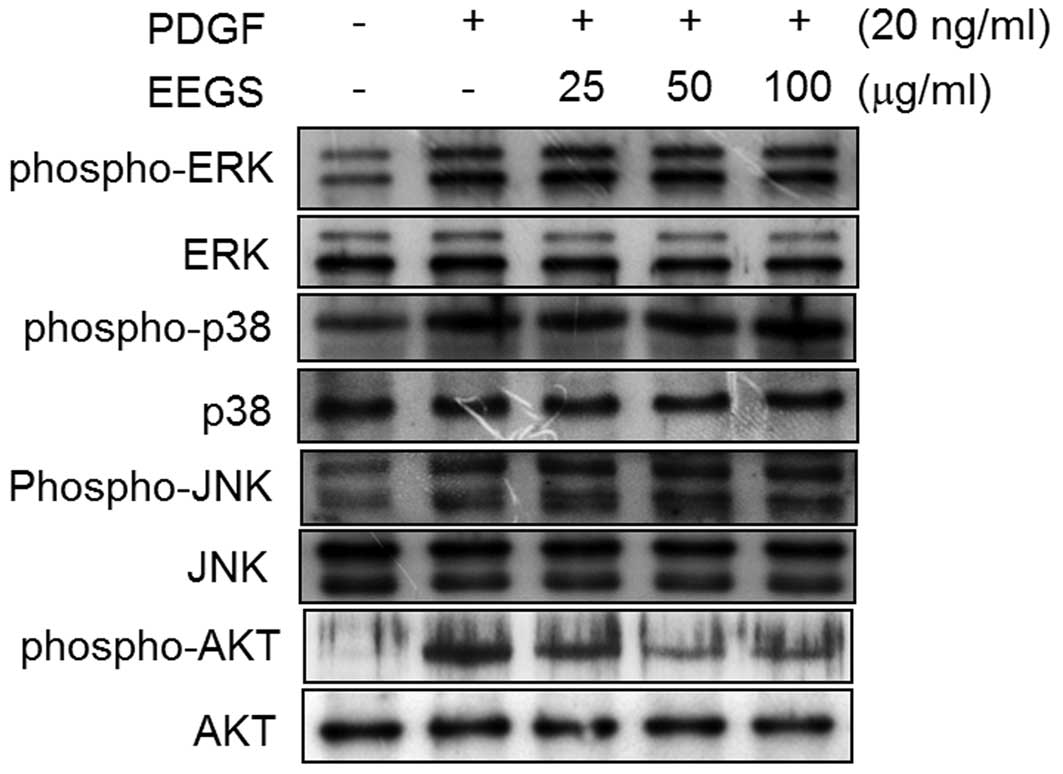 | Figure 3EEGS inhibits PDGF-induced
phosphorylation of Akt in VSMCs. Quiescent VSMCs were stimulated
with PDGF (20 ng/ml) in the presence or absence of the indicated
concentrations of EEGS for 10 min. Western blot analysis was
performed using antibodies specific for phospho-ERK1/2, ERK1/2,
phospho-p38, p38, phospho-JNK, JNK, phospho-Akt and Akt. PDGF,
platelet-derived growth factor; EEGS, ethanol extract of
Gleditsia sinensis thorns; VSMC, vascular smooth muscle
cell; ERK, extracellular signal-regulated kinase; JNK, c-Jun
N-terminal kinase; phospho, phosphorylated. |
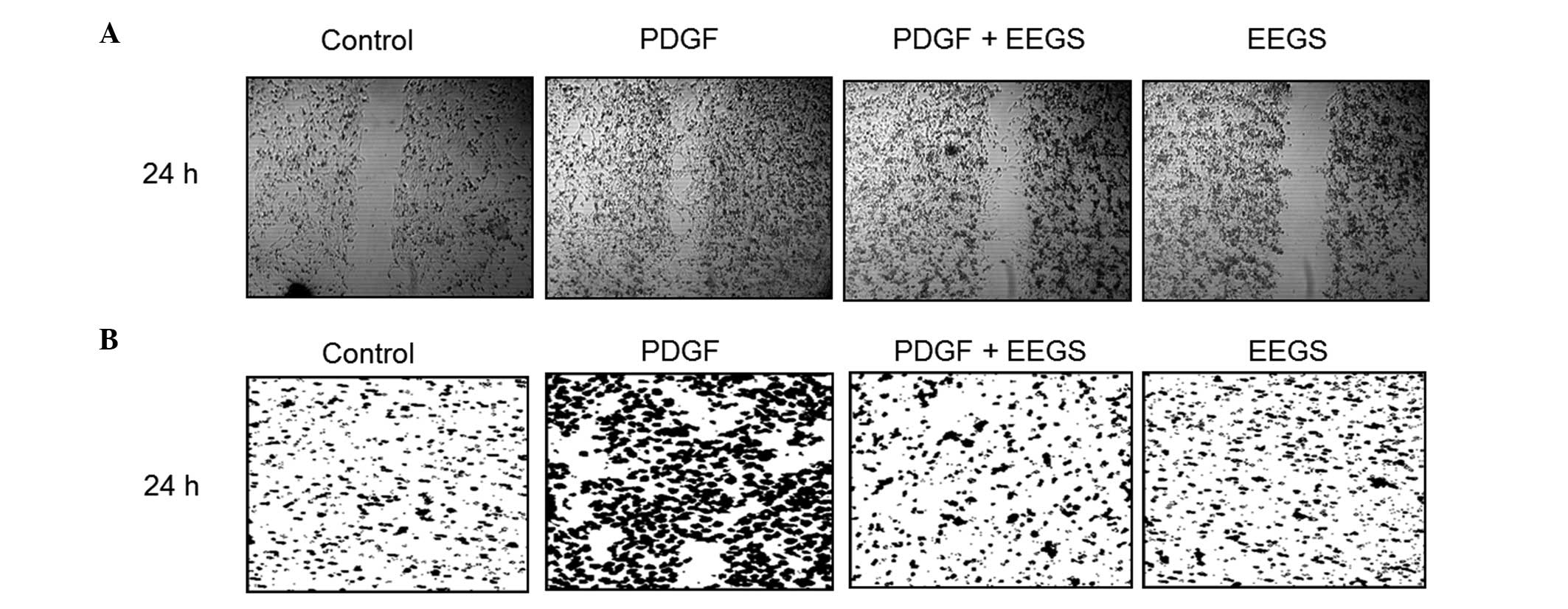
EEGS prevents the migration of
PDGF-induced VSMCs
The migration and invasion of VSMCs are highly
associated with the formation of vascular lesions (9). Therefore, wound-healing migration and
Matrigel™ invasion assays were used to investigate the role of EEGS
in the regulation of VSMC migration and invasion. Serum-starved
cells were wounded and incubated in the presence or absence of
PDGF. VSMC wounds were allowed to heal for 24 h subsequent to the
addition of PDGF. As shown in Fig.
4A, PDGF treatment was observed to significantly increase VSMC
migration. Furthermore, pretreatment with EEGS was found to
markedly inhibit the migration of VSMCs induced by PDGF at 24 h
(Fig. 4A). To confirm the
inhibitory effect of EEGS on PDGF-induced migration, a Matrigel
invasion assay was performed. PDGF treatment was associated with a
marked increase in VSMC invasiveness through the Matrigel-plated
Boyden chamber at 24 h, which was reduced with the addition of EEGS
(Fig. 4B). These results suggest
that EEGS is a potent inhibitor of PDGF-induced migration in
VSMCs.
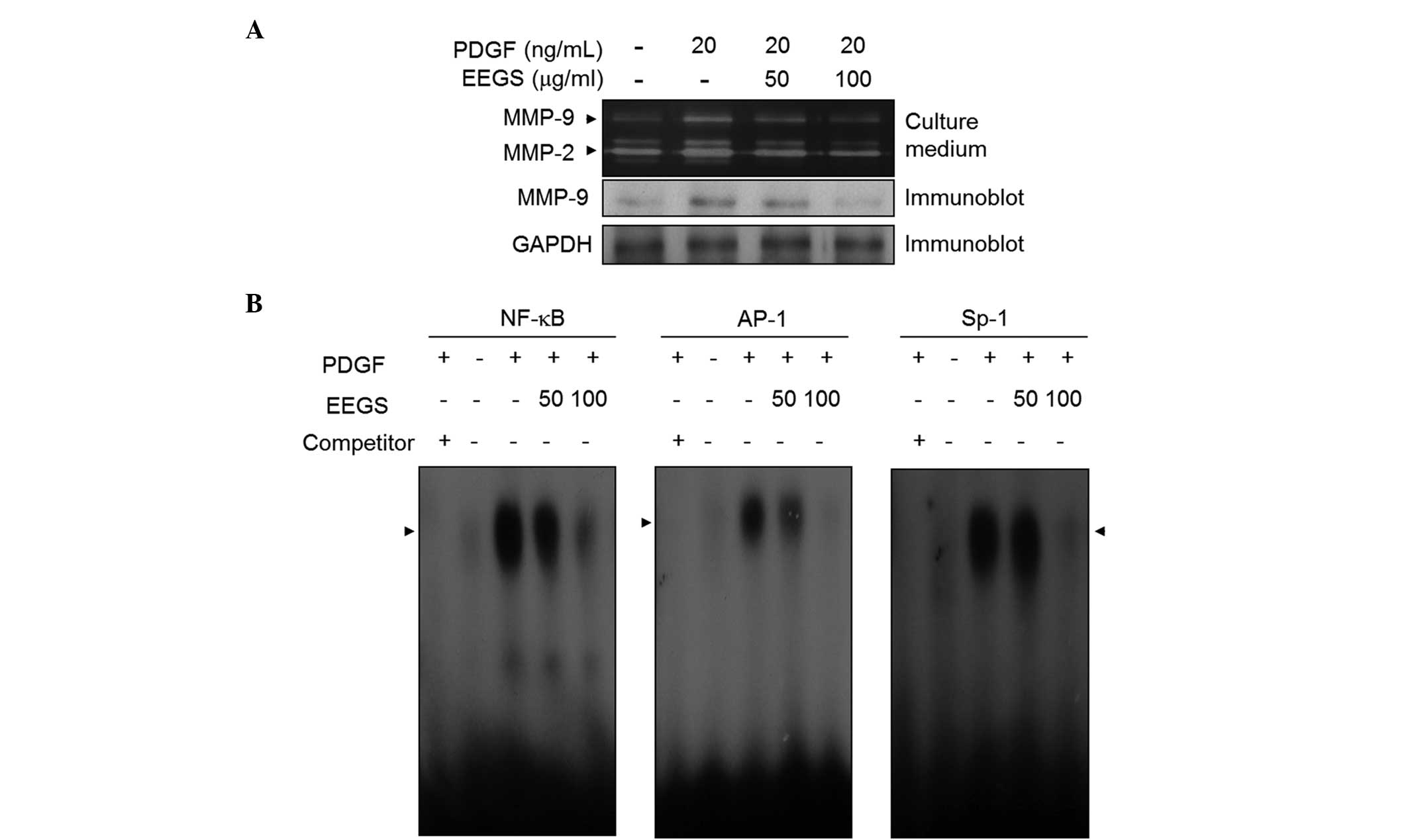
EEGS inhibits PDGF-stimulated MMP-9
expression through the suppression of NF-κB, AP-1 and Sp-1 binding
activities
MMP-9 expression is closely associated with VSMC
migration and invasion from media to intima (9–13).
In order to assess the role of EEGS in the expression of MMP-9, a
gelatin zymographic assay was performed. Media from VSMCs induced
by PDGF showed proteolytic activity at 92 kDa, corresponding to
MMP-9 (Fig. 5A). This induction of
MMP-9 expression by PDGF was suppressed following EEGS treatment
(Fig. 5A). Similar results were
observed with western blot analysis (Fig. 5A). Furthermore, under similar
experimental conditions, MMP-2 expression was inhibited by the
addition of EEGS to PDGF-treated VSMCs (Fig. 5A). To further understand the
mechanism underlying this suppressive effect on MMP-9 expression,
an EMSA assay was performed using three motifs: NF-κB, AP-1 and
Sp-1 cis-elements. Nuclear extracts from VSMCs treated with
PDGF markedly increased the binding activities of NF-κB, AP-1 and
Sp-1 (Fig. 5B). Furthermore, EEGS
treatment was observed to inhibit the increased binding activity of
the NF-κB, AP-1 and Sp-1 motifs (Fig.
5B). These data demonstrate that EEGS treatment is capable of
inhibiting MMP-9 expression, at least in part, by inhibiting the
binding activities of the NF-κB, AP-1 and Sp-1 transcription
factors.
Discussion
The thorns of Gleditsia sinensis have
demonstrated pharmacological effects on several systems, including
anti-cancer, anti-microbial and anti-inflammatory effects (17,20,21).
A previous study revealed the inhibitory effect of EEGS on the
proliferation of VSMCs (23).
However, the molecular mechanism of EEGS on PDGF-stimulated VSMC
responses is yet to be elucidated. The present study aimed to
investigate the precise mechanism of the anti-proliferative effect
of EEGS in PDGF-induced VSMCs. PDGF-induced VSMC proliferation has
a significant role in the pathogenesis of vascular lesion formation
(4,5).
Under normal conditions, VSMCs maintain a quiescent
status. However, subsequent to vascular injury, such as growth
factor stimulation, VSMCs re-enter the cell cycle in a state of
abnormal proliferation (1). It is
well established that PDGF is a key growth factor involved in VSMC
phenotypic change (4,5). In accordance with this, the present
study showed that PDGF induced the proliferation of VSMCs through
cell-cycle progression. Thus, in the present study, it was
hypothesized that EEGS was likely to inhibit cell proliferation
through the induction of cell-cycle arrest in PDGF-stimulated
VSMCs. As revealed by the MTT and thymidine uptake assays, EEGS
inhibited the PDGF-induced proliferation of VSMCs without
cytotoxicity in vitro. The inhibitory effect of EEGS was
associated with the accumulation of cells in the
G1-phase of the cell cycle. To further elucidate the
effect of EEGS on cell-cycle control, the role of cyclin-CDK
complexes, which induce a complex cascade of events (6–8)
during the induction of the G1-phase cell-cycle arrest,
was examined in the PDGF-stimulated VSMCs treated with EEGS. The
expression of cyclins and CDKs, cell-cycle regulatory proteins that
are essential for G1- to S-phase progression, was
investigated (6–8). The results of the present study
showed that EEGS treatment significantly downregulated cyclin D1,
cyclin E, CDK2 and CDK4 expression in PDGF-treated VSMCs. In
addition, the kinase activities associated with CDK2 and 4 were
inhibited by EEGS in PDGF-treated VSMCs. These results demonstrate
that the anti-proliferative effect of EEGS may be caused by the
G1-phase cell-cycle arrest associated with the
downregulation of cyclins and CDKs in PDGF-stimulated VSMCs.
The activity of cyclin/CDK complexes is highly
controlled by the binding of the CDKIs p21WAF1 and p27KIP1
(7,8). CDKIs bind tightly to the cyclin/CDK
complexes and inhibit their activity, resulting in an accumulation
of cells at the G1-phase boundary (7,8). It
has been previously reported that p27KIP1 expression is
downregulated following vascular injury (24). Furthermore, it has been shown that
overexpression of p27KIP1 has a suppressive effect on intimal
VSMCs, reducing neointimal hyperplasia in rat carotid arteries
(25). The present study showed an
inhibition of p27KIP1 expression in PDGF-treated VSMCs. This effect
was reversed following EEGS treatment, suggesting that the
EEGS-induced accumulation of p27KIP1 could be responsible for the
G1-phase arrest observed in the PDGF-treated VSMCs.
p21WAF1 was originally described as an inhibitor of cell
proliferation (7,8). However, several studies have
demonstrated that p21WAF1 is involved in VSMC proliferation
(26,27). In the present study, PDGF was found
to induce the expression of p21WAF1, and this effect was not
changed in VSMCs following EEGS treatment. These results
demonstrate that the G1-phase cell-cycle arrest induced
by EEGS is due to the decreased expression of cyclin/CDK complexes
through the induction of p27KIP1 in PDGF-treated VSMCs.
MAPK and Akt are closely implicated in VSMC
proliferation induced by mitogenic stimuli, such as PDGF (4,5).
Therefore, the effect of the early signal transduction pathway in
response to PDGF stimulation in VSMCs was examined. PDGF treatment
has previously been demonstrated to induce the phosphorylation of
Akt and MAPKs, including ERK1/2, JNK and p38MAPK, in VSMCs
(4,5). In accordance with these previous
findings, EEGS treatment in the present study was found to
significantly attenuate PDGF-induced Akt phosphorylation in VSMCs.
However, EEGS treatment was not observed to affect the
phosphorylation of MAPKs, including ERK1/2, JNK and p38MAPK, in
PDGF-treated VSMCs. These results suggest that EEGS inhibited
PDGF-stimulated proliferation in VSMCs through the suppression of
Akt phosphorylation. Although a previous study has reported that
the phosphorylation of MAPKs, including ERK1/2, JNK and p38MAPK, is
induced following EEGS treatment in colon cancer cells (21), to the best of our knowledge, this
is the first study to show that the suppression of Akt
phosphorylation is involved in the EEGS-induced inhibition of cell
proliferation.
It is well established that the migration and
invasion of VSMCs have a role in atherosclerotic lesion formation
(1,3,9).
Several studies have demonstrated that cytokines and growth
factors, produced by various types of stimuli, promote the invasion
and migration of VSMCs (1,3,9). The
enhanced invasive and migratory capacity of VSMCs has been
associated with the presence of the growth factor PDGF (1,9). In
the present study, a wound-healing assay revealed an upregulation
in migratory potential in VSMCs treated with PDGF. Similar results
were observed using a Matrigel invasion assay. Of note, the
significant reduction in the migratory and invasive capacity
observed in PDGF-stimulated VSMCs treated with EEGS was not a
consequence of cell viability. These results demonstrate that EEGS
may be an inhibitor of the migration and invasion that is induced
by PDGF in VSMCs.
VSMC migration requires degradation of the ECM,
which results in the formation of neointimal lesions. The primary
mechanism involved in VSMC migration is the production of
proteolytic enzymes, including MMP-2 and -9 (9–13). A
growing body of evidence suggests that MMP-9 expression may
contribute to the enhanced progression of arterial vascular lesions
(9–13). Therefore, in the present study,
MMP-9 expression was assessed in PDGF-treated VSMCs. In accordance
with the present migration and invasion results, MMP-9 expression
was increased with PDGF treatment. To further elucidate the
regulation of MMP-9 expression by PDGF in VSMCs, an EMSA assay was
performed. In the present study, the transcription factors NF-κB,
AP-1 and Sp-1 were identified to be involved in MMP-9 expression in
PDGF-stimulated VSMCs. Therefore, NF-κB, AP-1 and Sp-1 may
co-operate in the activation of the MMP-9 gene in PDGF-treated
VSMCs. Next, the effect of EEGS treatment on the inhibition of
MMP-9 expression was examined in PDGF-induced VSMCs. EEGS was
observed to reduce PDGF-stimulated MMP-9 expression in VSMCs, as
determined by zymography and western blot analyses. Furthermore, an
EMSA assay using consensus NF-κB, AP-1 and Sp-1 probes showed that
EEGS treatment induced a significant decrease in the binding
activities of NF-κB, AP-1 and Sp-1 in PDGF-treated VSMCs. These
results showed that the ability of EEGS to inhibit MMP-9 expression
in PDGF-treated VSMCs may be achieved through the suppression of
NF-κB, AP-1 and Sp-1 binding activities.
The accumulation of VSMCs in the arterial intima is
a key event in the pathogenesis of cardiovascular diseases, which
is characterized by the formation of neointima resulting from the
proliferation and migration of VSMCs from media to intima (1). A proliferating and migrating cell
population and increased PDGF expression are observed in the
neointimal layer of severely injured arteries (28), suggesting that the PDGF-induced
proliferation and migration of VSMCs are important features in
neointimal formation following vascular injury, and may lead to
cardiovascular disease (4,5,28).
Results from the present study demonstrated that EEGS treatment
significantly inhibited proliferation and migration in
PDGF-stimulated VSMCs, without cell toxicity. The proliferation and
migration of VSMCs is a key factor in neointimal formation;
therefore, the results of this study suggest that an additional
mechanism exists by which EEGS treatment may be critical in
preventing the progression of atherosclerosis and restenosis.
In conclusion, to the best of our knowledge, the
present study has provided the first evidence that EEGS inhibits
the proliferation of PDGF-simulated VSMCs via Akt phosphorylation
without cell death. EEGS also induced G1-phase
cell-cycle arrest, as a result of the decreased levels of cyclin
D1/CDK4 and cyclin E/CDK2 that were mediated by the upregulation of
p27KIP1 expression. In addition, EEGS treatment was found to
suppress migration and invasion in PDGF-stimulated VSMCs.
Furthermore, EEGS markedly reduced the PDGF-induced expression of
MMP-9 through the suppression of NF-κB, AP-1 and Sp-1 binding
activities. The results of the present study may, in part, explain
the therapeutic potential of EEGS for the prevention of
cardiovascular diseases associated with multiple pathological
events involving the proliferation and migration of VSMCs.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF), funded by the Ministry of Education Science and Technology
(grant no. 2008-0062611). This study was also supported by the
Chung-Ang University research grants 2013.
References
|
1
|
Ross R: Cell biology of atherosclerosis.
Annu Rev Physiol. 57:791–804. 1995. View Article : Google Scholar
|
|
2
|
Dzau VJ, Braun-Dullaeus RC and Sedding DG:
Vascular proliferation and atherosclerosis: new perspectives and
therapeutic strategies. Nat Med. 8:1249–1256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar
|
|
4
|
Heldin CH and Westermark B: Mechanism of
action and in vivo role of platelet-derived growth factor. Physiol
Rev. 79:1283–1316. 1999.PubMed/NCBI
|
|
5
|
Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao
T, Miyazaki H and Iwao H: Role of JNK, p38, and ERK in
platelet-derived growth factor-induced vascular proliferation,
migration, and gene expression. Arterioscler Thromb Vasc Biol.
23:795–801. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sherr CJ: G1 phase progression: cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Newby AC and Zaltsman AB: Molecular
mechanisms in intimal hyperplasia. J Pathol. 190:300–309. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cho A and Reidy MA: Matrix
metalloproteinase-9 is necessary for the regulation of smooth
muscle cell replication and migration after arterial injury. Circ
Res. 91:845–851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galis ZS, Johnson C, Godin D, Magid R,
Shipley JM, Senior RM and Ivan E: Targeted disruption of the matrix
metalloproteinase-9 gene impairs smooth muscle cell migration and
geometrical arterial remodeling. Circ Res. 91:852–859. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho A, Graves J and Reidy MA:
Mitogen-activated protein kinases mediate matrix
metalloproteinase-9 expression in vascular smooth muscle cells.
Arterioscler Thromb Vasc Biol. 20:2527–2532. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moon SK, Cha BY and Kim CH: ERK1/2
mediates TNF-alpha-induced matrix metalloproteinase-9 expression in
human vascular smooth muscle cells via the regulation of NF-kappaB
and AP-1: Involvement of the ras dependent pathway. J Cell Physiol.
198:417–427. 2004. View Article : Google Scholar
|
|
14
|
Dollery CM, McEwan JR and Henney AM:
Matrix metalloproteinases and cardiovascular disease. Circ Res.
77:863–868. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hansson GK and Robertson AK: TGF-beta in
atherosclerosis. Arterioscler Thromb Vasc Biol. 24:E1372004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lim JC, Park JH, Budesinsky M, Kasal A,
Han YH, Koo BS, Lee SI and Lee DU: Antimutagenic constituents from
the thorns of Gleditsia sinensis. Chem Pharm Bull (Tokyo).
53:561–564. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou L, Li D, Wang J, Liu Y and Wu J:
Antibacterial phenolic compounds from the spines of Gleditsia
sinensis Lam. Nat Prod Res. 21:283–291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li WH, Zhang XM, Tian RR, Zheng YT, Zhao
WM and Qiu MH: A new anti-HIV lupane acid from Gleditsia
sinensis Lam. J Asian Nat Prod Res. 9:551–555. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin TY and Kim DK: Inhibitory effect of
mast cell-dependent anaphylaxis by Gleditsia sinensis. Arch
Pharm Res. 23:401–406. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park E and Shin MJ: Anti-inflammatory
activity of aqueous extract from Gleditsiae Spina. J
Pharmaceut Soc Korea. 37:124–128. 1993.
|
|
21
|
Lee SJ, Cho YH, Kim H, Park K, Park SK, Ha
SD, Kim WJ and Moon SK: Inhibitory effects of the ethanol extract
of Gleditsia sinensis thorns on human colon cancer HCT116
cells in vitro and in vivo. Oncol Rep. 22:1505–1512.
2009.PubMed/NCBI
|
|
22
|
Zhou L, Li D, Jiang W, Qin Z, Zhao S, Qiu
M and Wu J: Two ellagic acid glycosides from Gleditsia
sinensis Lam. with antifungal activity on Magnaporthe
grisea. Nat Prod Res. 21:303–309. 2007.
|
|
23
|
Lee SJ, Park SS, Kim WJ and Moon SK:
Gleditsia sinensis thorn extract inhibits proliferation and
TNF-α-induced MMP-9 expression in vascular smooth muscle cells. Am
J Chin Med. 40:373–386. 2012. View Article : Google Scholar
|
|
24
|
Tanner FC, Yang ZY, Duckers E, Gordon D,
Nabel GJ and Nabel EG: Expression of cyclin-dependent kinase
inhibitors in vascular disease. Circ Res. 82:396–403. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen D, Krasinski K, Sylvester A, Chen J,
Nisen PD and Andrés V: Downregulation of cyclin-dependent kinase 2
activity and cyclin A promoter activity in vascular smooth muscle
cells by p27(KIP1), an inhibitor of neointima formation in the rat
carotid artery. J Clin Invest. 99:2334–2341. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moon SK, Kim HM, Lee YC and Kim CH:
Disialoganglioside (GD3) synthase gene expression suppresses
vascular smooth muscle cell responses via the inhibition of ERK1/2
phosphorylation, cell cycle progression, and matrix
metalloproteinase-9 expression. J Biol Chem. 279:33063–33070. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shankland SJ and Wolf G: Cell cycle
regulatory proteins in renal disease: role in hypertrophy,
proliferation, and apoptosis. Am J Physiol Renal Physiol.
278:F515–F529. 2000.PubMed/NCBI
|
|
28
|
Uchida K, Sasahara M, Morigami N, Hazama F
and Kinoshita M: Expression of platelet-derived growth factor
B-chain in neointimal smooth muscle cells of balloon injured rabbit
femoral arteries. Atherosclerosis. 124:9–23. 1996. View Article : Google Scholar : PubMed/NCBI
|