Introduction
It is well known that a lethal ventricular
tachyarrhythmia, including ventricular fibrillation, is one of the
most common reasons for sudden cardiac death following myocardial
infarction (MI). Multiple factors may contribute to the genesis of
ventricular arrhythmia (VA); the electrical remodeling generates a
proarrhythmogenic substrate following MI. The proarrhythmogenic
substrate may be involved in altered ionic currents, action
potentials, cardiac fibrosis and cell-to-cell coupling. Therefore,
novel approaches that prevent electrical remodeling following MI
are required.
Artemisinin is the active component of Artemisia
annua L. and is approved worldwide for the treatment and
prevention of malaria (1). In
addition to its antimalarial properties, previous studies have
demonstrated that artemisinin significantly inhibits ventricular
remodeling. The present study demonstrated that artemisinin
attenuates ventricular remodeling and neural remodeling following
MI by exhibiting anti-inflammatory effects (2,3).
However, whether artemisinin is able to attenuate electrical
remodeling following MI remains unclear. In order to assess our
hypothesis, a rat MI model was used to determine whether
artemisinin prevents electrical remodeling following MI.
Materials and methods
Animal preparation
All experiments were approved by the Institutional
Animal Care and Use Committee of Wuhan University (Wuhan, China)
and were conducted in accordance with the Guideline for the Care
and Use of Laboratory Animals.
Adult male Sprague-Dawley rats (Center for Animal
Experiments, Wuhan University, Wuhan, China) weighing 250–300 g
were anesthetized with an intraperitoneal injection of 3%
pentobarbital sodium (30 mg/kg). A left intercostal thoracotomy was
performed to expose the heart and the left anterior descending
(LAD) artery was ligated at the origin. The sham group underwent
thoracotomy and pericardiotomy; however, not LAD ligation (4). The chest was then closed and the
animals were allowed to recover in a warm, clean cage. Fatalities
within 24 h of surgery were excluded from the present study.
Reagents
Artemisinin was purchased from the Guilin
Pharmaceutical Factory (Guangxi, China). The drug was dissolved in
0.5% carboxymethyl cellulose (CMC) immediately prior to use. The
drug safety of artemisinin up to 28 days was presented in a
previous study (5). The vehicle
(0.5% CMC) was used as a control.
Treatment protocol
The rats surviving 24 h following MI were randomly
assigned to two treatment groups as follows: 75 mg/kg/day
artemisinin by oral gavage three times a day for four weeks (MI +
artemisinin group; n=21); the same volume of 0.5% CMC liquid
vehicle by oral gavage three times a day for four weeks after MI
(MI + vehicle group; n=21) and the same treatment as the MI +
vehicle group in the sham operation group 24 h after sham operation
(thoracotomy with LAD isolation, however, without ligation; n=10)
for four weeks.
Measurement of tumor necrosis factor
(TNF)-α levels
Blood (0.75 ml) was collected into chilled EDTA
tubes one day prior to MI surgery and subsequently on days 1, 3, 5,
7, 14, 21 and 28 after surgery. The blood samples were centrifuged
at 1,100 × g, 4°C and the plasma samples were separated and stored
at -70°C until they were assayed for TNF-α. Red blood cells were
suspended in an equal volume of heparinized saline and
reinfused.
The TNF-α levels in the rats were measured using an
ultrasensitive rat TNF-α ELISA kit (Biosource International, Inc.,
Camarillo, CA, USA) according to the manufacturer’s instructions.
The details of the methodology were described in a previous study
(6).
Electrophysiological evaluation
To assess the inducibility of VAs, the burst stimuli
was used (2 ms pulses at 50 Hz, 2 sec burst duration) and burst
pacing was used for up to 3 min of pacing in the infarcted border
zone (IBZ).
To assess the ventricular fibrillation threshold
(VFT), electrical stimulation was supplied with 100 Hz to the right
ventricle. Each stimulation was administered for 30 sec. The
interval between each episode of stimulation was 1 min. The initial
pacing voltage was 1 V and progressively increased by 0.5 V. VFT
was defined as the lowest voltage at which ventricular fibrillation
was induced and sustained for at least 20 sec (7).
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted from the non-infarcted zone
(NIZ) and the IBZ of the left ventricle using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. qPCR was performed using the ABI Prism
7000 (Applied Biosystems, Foster City, CA, USA). cDNA was amplified
under the following conditions: 94°C for 10 min and then for 45
cycles at 94°C for 10 sec and 56°C for 30 sec. The primer sequences
of connexin 43 (Cx43) and glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) are shown in Table I. For
quantification, Cx43 expression was normalized to GAPDH. The
reactions were programmed on a computer linked to the detector (ABI
Prism 7000 Sequence Detection system; Applied Biosystems) for 40
cycles of the amplification step. Experiments were replicated three
times and the results are expressed as the mean value.
 | Table IPrimer sequences for Cx43 and
actin. |
Table I
Primer sequences for Cx43 and
actin.
| Gene | Primer sequence |
|---|
| Cx43 |
| Forward |
ACAGCGCAGAGCAAAATCG |
| Reverse |
ATGGCTGGAGTTCATGTCCAG |
| Actin |
| Forward | GCTCCTCCTG
AGCGCAAGTA |
| Reverse |
CCTGCTTGCTGATCCACATCT |
Western blot analysis of Cx43
The peri-infarcted zone of the left ventricle and
the same zone of the left ventricle in the sham group were used for
western blot analysis. Equal quantities of protein were loaded and
separated by SDS-PAGE, transferred onto a nitrocellulose membrane
and incubated with primary antibodies (anti-Cx43, polyclonal
antibody; dilution 1:1,000; Abcam, Cambridge, MA, USA) and
anti-GAPDH (1:2,000; Abcam) overnight at 4°C. Horseradish
peroxidase-conjugated anti-rabbit immunoglobulin IgG (1:1,500;
Beyotime Institute of Biotechnology, Shanghai, China) was applied
as the secondary antibody for 1 h at room temperature. Finally, the
blots were visualized using an enhanced chemiluminescence kit
(Beyotime Institute of Biotechnology) and the signals were analyzed
using a Bio-Rad image system (Bio-Rad, Hercules, CA, USA).
Immunofluorescent studies of Cx43
Immunofluorescence staining was performed to
investigate the localization and the distribution of Cx43 at the
peri-infarct zone of the left ventricle. Fresh tissue was fixed in
4% paraformaldehyde for 24 h, then dehydrated in 30% sucrose for 48
h. The tissue was then embedded at −25°C (frozen section inside the
machine) and then frozen sectioning was performed. Following
inhibition with goat serum for 30 min, the frozen slices (5 μm
thick) were incubated with rabbit polyclonal anti-Cx43 antibody
(Zymed Life Technologies, Carlsbad, CA, USA) overnight at 4°C and
then incubated with fluorescein isothiocyanate (FITC)-conjugated
anti-rabbit IgG (Zymed Life Technologies) for 1 h at room
temperature. The specimens were examined under a Leica M205 FA
fluorescence microscope (Leica, Wetzlar, Germany).
Statistical analysis
All values are expressed as the mean ± standard
deviation. Comparisons between groups were performed using one way
analysis of variance and the least significant difference test was
used for post hoc multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
Animal survival rate and infarct
size
At 28 days after MI, the survival rate in the
artemisinin treatment group (68.88%; 31/45) was significantly
higher than that in the vehicle-treated group (42.22%, 19/45;
P<0.05). The infarct size four weeks after MI was similar
between the MI + artemisinin group and the MI + vehicle group
(34.32±1.68% and 33.18±1.42%, respectively; P>0.05).
Plasma TNF-α levels
As shown in Fig. 1,
the baseline plasma TNF-α level (pg/ml) in the three groups
observed were comparable across groups (P>0.05). In the sham
group, TNF-α levels remained at or around baseline during the
entire study. By contrast, in the MI + vehicle group the TNF-α
level was increased within 24 h after MI (25.6±4.2) and remained
elevated (26.71±4.11, 46.18±4.7, 56.42±3.47, 117.52±8.72,
150.8±11.31, 191.08±12.68 on days 3, 5, 7, 14, 21, 28 after MI,
respectively) throughout the study. The TNF-α level in the MI +
artemisinin group also increased within 24 h (24.46±6.15), however,
decreased (18.24±3.24) on day 3, returned to baseline by day 5 and
then remained low (10.31±3.09, 7.72±2.08, 9.95±2.37, 8.08±2.49,
10.33±3.08) throughout the study.
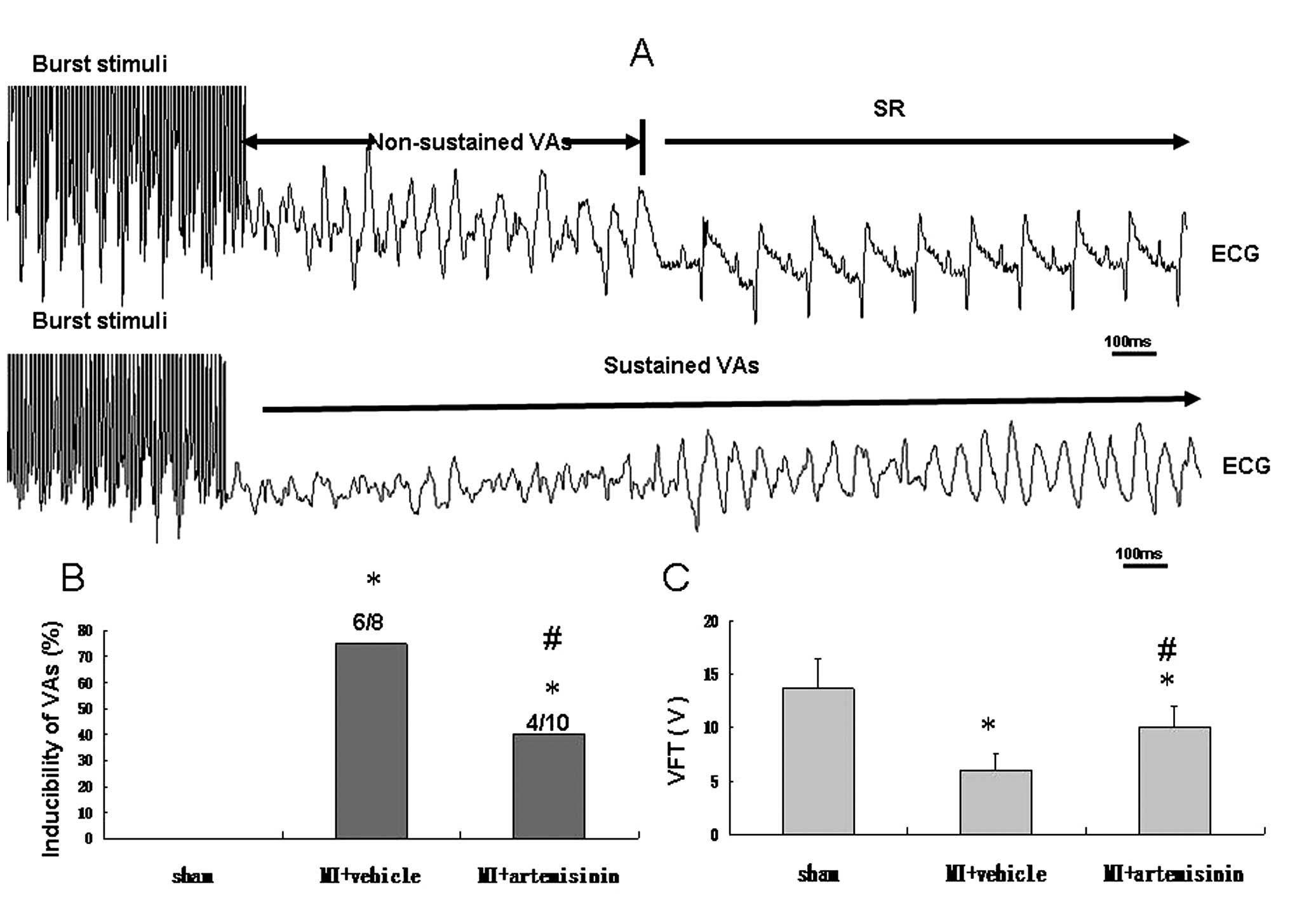
Inducibility of VAs and VFT
The VA was induced by burst stimuli, compared with
the sham group, the inducibility was significantly higher in the MI
+ vehicle group; however, the inducibility was reduced in the
artemisinin treatment group (P<0.05). VFT, reflects the
vulnerability of ventricular fibrillation and VFT was significantly
decreased in the MI + vehicle group and only marginally decreased
in the MI + artemisinin group (Fig.
2).
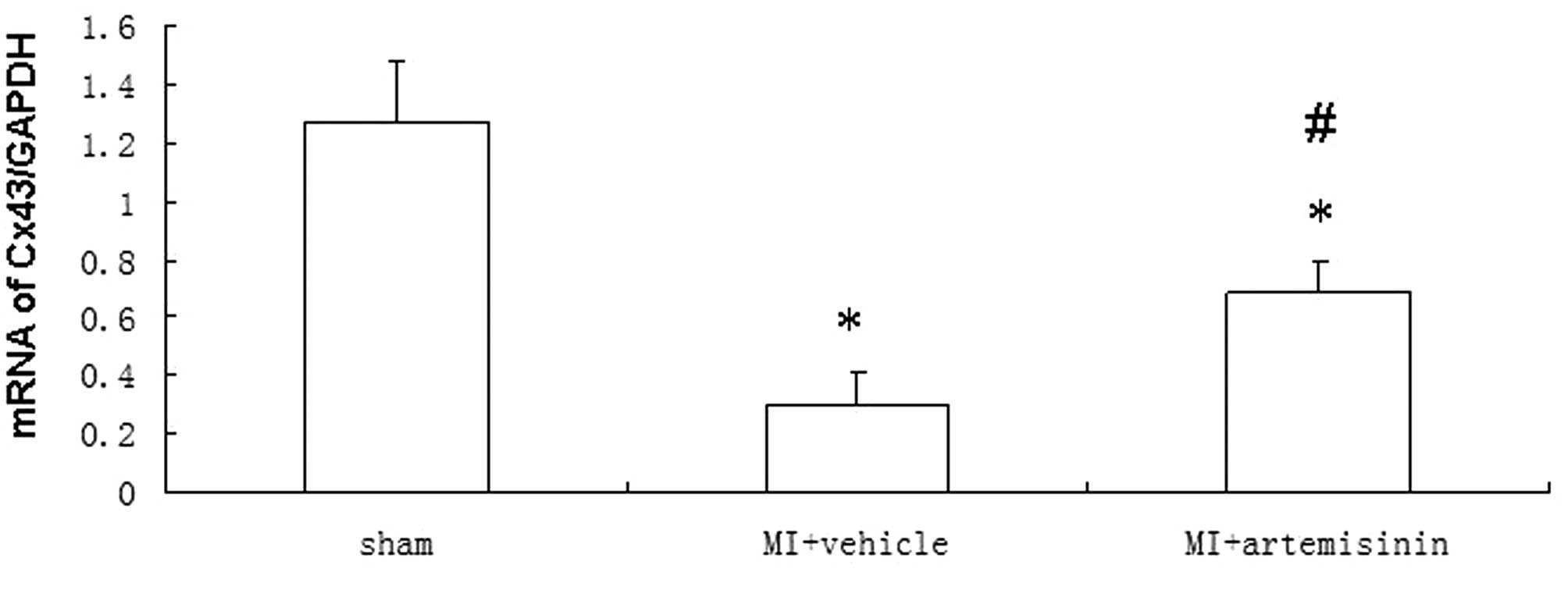
qPCR of Cx43
The Cx43 mRNA levels 4 weeks after MI demonstrated a
significant downregulation at the IBZ in the vehicle-treated group
compared with the sham group. However, the reduction of Cx43 mRNA
expression in the artemisinin-treated group following MI was
significantly smaller than the reduction observed in the
vehicle-treated group (Fig.
3).
Western blot analysis
The level of Cx43 protein was investigated by
western blotting. Representative blots and quantitative results are
presented in which Cx43 band intensities are normalized to β-actin.
Two predominant forms of Cx43 were detected, the phosphorylated
form of Cx43 (P-Cx43; 43 kDa) and the non-phosphorylated form of
Cx43 (NP-Cx43; 41 kDa). Compared with the sham group, the
P-Cx43/β-actin was significantly reduced in the MI + vehicle group.
By contrast, the decrease in P-Cx43/β-actin was reversed in the MI
+ artemisinin group (Fig. 4).
Immunofluorescent studies of Cx43
The connexin localization was evaluated using an
immunofluorescent technique. Cx43 protein was only located at the
intercalated disk area in the left ventricle of the sham group or
normal tissue far from the infarcted zone. However, the
distribution of Cx43 on the IBZ was disrupted. In the areas of the
IBZ, Cx43 was distributed on the lateral side of the myocyte,
however, not in the intercalated disk area. The disarray of Cx43
was ameliorated in the IBZ of the artemisinin treatment group,
where its distribution was relatively normal. Quantitative analysis
of the Cx43-signal intensity demonstrated that the Cx43-signal in
the vehicle-treated group was markedly reduced in the IBZ, however,
was partially attenuated by artemisinin treatment (Fig. 5).
 | Figure 5Immunofluorescent studies of Cx43
following MI. (A) Cx43 signals were mainly located at end-to-end
apposition among the neighboring cells in the sham group. By
contrast, MI markedly decreased the expression of Cx43 in the IBZ;
however, not in the NIZ. Following administration of artemisinin,
Cx43 signals were increased in the IBZ. Blue fluorescence indicates
nuclei (DAPI) and red indicates Cx43 (rabbit polyclonal anti-Cx43
antibody). (B) Proportion of the total cell area occupied by Cx43
immunoreactive signals at the border zone. Data are expressed as
the mean ± standard deviation. *P<0.05, compared with
the sham group, #P<0.05, compared with the MI +
vehicle group. Cx43, connexin 43; MI, myocardial infarction; IBZ,
infarcted border zone; NIZ, non-infarcted zone. |
Discussion
To the best of our knowledge, the present study
demonstrated for the first time that artemisinin is able to
ameliorate electrical remodeling at the IBZ following MI. Firstly,
artemisinin treatment significantly inhibited the reduction of
total Cx43 and phosphorylated Cx43. Secondly, the ischemia-induced
disarrangement and distribution were reversed following artemisinin
treatment. Thirdly, artemisinin treatment reduced the inducibility
of VAs and increased VFT in rats with MI.
Artemisinin is the active component of Artemisia
annua L. and is approved worldwide for the treatment and
prevention of malaria (1).
Previous studies have indicated that artemisinin is important in
cardioprotection, and that it is able to attenuate ventricular
remodeling and neural remodeling (2–3). The
present study demonstrated the anti-arrhythmic effect of
artemisinin.
It is well known that Cx43, which is the predominant
ventricular gap junction protein, is critical for maintaining
normal cardiac electrical conduction. Cardiac-restricted knockout
of Cx43 causes a slowed ventricular conductive velocity and
spontaneous VAs (8–9). Apart from reducing conduction, Cx43
remodeling may be associated with action potential duration
dispersion in the failing heart (10). Furthermore, increased Cx43 by
either Cx43 gene transfer or transplantation of embryonic
cardiomyocytes decreases the spatial heterogeneity of
repolarization and has the potential to reduce life-threatening
post-infarct arrhythmias (11–12).
Post-MI, the epicardial border zone (EBZ) demonstrated a marked
disruption in gap-junctional distribution, with Cx43 disarrayed
along the lateral surfaces of the cells (13) and selectively reduced in the EBZ
(14). The present study observed
that the disarray of Cx43 was ameliorated in the IBZ of the
artemisinin-treated heart, displaying a relatively normal
distribution. In addition, the present study demonstrated that the
Cx43 mRNA level and the quantity of Cx43 protein were significantly
increased at the IBZ by artemisinin therapy. The beneficial effects
on Cx43 may improve electrical conduction and ameliorate
repolarization dispersion in the IBZ. It may provide a rational
explanation for why artemisinin prevented the induction of VAs in
the present study.
Although the present study suggests that the
mechanisms by which artemisinin produces anti-arrhythmic effects
are associated with increases in Cx43 expression, other potential
mechanisms require investigation. Firstly, our previous studies
demonstrated that artemisinin attenuates interstitial fibrosis
(2) and sympathetic reinnervation
in the IBZ (3), which contributes
to VAs and sudden cardiac death. Secondly, previous studies have
confirmed that artemisinin exerts a direct effect on the ion
channels (15–16). Therefore, it was hypothesized that
artemisinin produces anti-arrhythmic effects by directly regulating
the cardiac ionic channel.
Several potential mechanisms may be involved in the
ability of artemisinin to inhibit Cx43 degradation and increase
Cx43 expression following MI. It has been demonstrated that
myocardial ischemia with sympathetic nerve stimulation promoted the
degradation of the Cx43 protein (17) and vagal nerve stimulation protects
the heart against ischemic-induced arrhythmias by preserving Cx43
protein (18), suggesting that
autonomic nerve activity correlates with the survival time of Cx43.
Our previous study revealed that artemisinin inhibits neural
remodeling and sympathetic hyperinnervation following MI in rats
(3). One mechanism by which
artemisinin inhibits the degradation of Cx43 following MI may be by
directly attenuating sympathetic tone. Secondly, artemisinin is
able to upregulate the mRNA level of Cx43, indicating that
increasing Cx43 protein at the transcriptional level may serve as
another mechanism. Thirdly, inflammatory factors, including TNF-α,
which is able to aggravate the dephosphorylation and degradation of
Cx43 (19), were increased
significantly following MI. Artemisinin is able to inhibit the
levels of TNF-α following MI, which contributes to the upregulation
of the Cx43 protein.
There are several limitations in the present study.
Firstly, the transmural heterogeneity of Cx43 expression was not
evaluated in the present study, such an analysis is required to
clarify the precise mechanisms for the anti-arrhythmic effects of
artemisinin. Secondly, although artemisinin was demonstrated to
attenuate the reduction of Cx43 possibly by reducing TNF-α levels
in the IBZ, the present study did not provide direct evidence by
evaluating the effect of artemisinin on Cx43 expression following
inhibition of TNF-α expression. Finally, the cardiac morphology and
hemodynamics were not measured at the end of the study, although
the results were demonstrated in our previous study (2).
In conclusion, artemisinin reduces the vulnerability
to VAs and increases the VFT following MI. The cardioprotective
effects of artemisinin may occur by reducing TNF-α levels and
preventing gap junction remodeling. The results suggest that
artemisinin is a potential therapeutic candidate for the prevention
of ventricular electrical remodeling following MI.
Acknowledgements
This study was supported by the Fundamental Research
funds for the Central Universities (grant no. 201130202020003), the
Natural Science Foundation of Hubei province, China (grant no.
2011CHB034), the National Natural Science Foundation of China
(grant no. 81270305) and the National Science & Technology
Pillar Program of China (grant no. 2011BAI11B12).
References
|
1
|
Haynes RK: Artemisinin and derivatives:
the future for malaria treatment? Curr Opin Infect Dis. 14:719–726.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gu Y, Wang X, Wang X, Yuan M, Wu G, Hu J,
Tang Y and Huang C: Artemisinin attenuates post-infarct myocardial
remodeling by down-regulating the NF-κB pathway. Tohoku J Exp Med.
227:161–170. 2012.PubMed/NCBI
|
|
3
|
Gu Y, Wang X, Wu G, Wang X, Cao H, Tang Y
and Huang C: Artemisinin suppresses sympathetic hyperinnervation
following myocardial infarction via anti-inflammatory effects. J
Mol Histol. 43:737–743. 2012. View Article : Google Scholar
|
|
4
|
Wen HZ, Jiang H, Li L, Xie P, Li JY, Lu Zb
and He B: Semaphorin 3A attenuates electrical remodeling at infarct
border zones in rats after myocardial infarction. Tohoku J Exp Med.
225:51–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiong Z, Sun G, Zhu C, Cheng B, Zhang C,
Ma Y and Dong Y: Artemisinin, an anti-malarial agent, inhibits rat
cardiac hypertrophy via inhibition of NF-κB signaling. Eur J
Pharmacol. 649:277–284. 2010.PubMed/NCBI
|
|
6
|
Ponnappa BC, Dey I, Tu GC, Zhou F, Aini M,
Cao QN and Israel Y: In vivo delivery of antisense oligonucleotides
in pH-sensitive liposomes inhibits lipopolysaccharide-induced
production of tumor necrosis factor-α in rats. J Pharmacol Exp
Ther. 297:1129–1136. 2001.PubMed/NCBI
|
|
7
|
Wen H, Jiang H, Lu Z, He B, Hu X, Chen J
and Zhao D: Carvedilol ameliorates the decreases in connexin 43 and
ventricular fibrillation threshold in rats with myocardial
infarction. Tohoku J Exp Med. 218:121–127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lerner DL, Yamada KA, Schuessler RB and
Saffitz JE: Accelerated onset and increased incidence of
ventricular arrhythmias induced by ischemia in Cx43-deficient mice.
Circulation. 101:547–552. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao JA, Gutstein DE, Liu F, Fishman GI and
Wit AL: Cell coupling between ventricular myocyte pairs from
connexin43-deficient murine hearts. Circ Res. 93:736–743. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Poelzing S and Rosenbaum DS: Altered
connexin43 expression produces arrhythmia substrate in heart
failure. Am J Physiol Heart Circ Physiol. 287:H1762–H1770. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Amino M, Yoshioka K, Tanabe T, Tanaka E,
Mori H, Furusawa Y, et al: Heavy ion radiation up-regulates Cx43
and ameliorates arrhythmogenic substrates in hearts after
myocardial infarction. Cardiovasc Res. 72:412–421. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fernandes S, van Rijen HV, Forest V, Evain
S, Leblond AL, Mérot J, et al: Cardiac cell therapy: overexpression
of connexin43 in skeletal myoblasts and prevention of ventricular
arrhythmias. J Cell and Mol Med. 13:3703–3712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peters NS, Coromilas J, Severs NJ and Wit
AL: Disturbed connexin43 gap junction distribution correlates with
the location of reentrant circuits in the epicardial border zone of
healing canine infarcts that cause ventricular tachycardia.
Circulation. 95:988–996. 1997. View Article : Google Scholar
|
|
14
|
Ohara T, Ohara K, Cao JM, et al: Increased
wave break during ventricular fibrillation in the epicardial border
zone of hearts with healed myocardial infarction. Circulation.
103:1465–1472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiao G, Li S, Yang B and Li B: Inhibitory
effects of artemisinin on voltage-gated ion channels in intact
nodose ganglion neurones of adult rats. Basic Clin Pharmacol
Toxicol. 100:217–224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang BF, Luo DL, Bao LH, Zhang YC and Wang
HZ: Artemisinin blocks activating and slowly activating
K+ current in guinea pig ventricular myocytes. Zhongguo
Yao Li Xue Bao. 19:269–272. 1998.PubMed/NCBI
|
|
17
|
Jiang H, Hu X, Lu Z, Wen H, Zhao D, Tang
Q, et al: Effects of sympathetic nerve stimulation on
ischemia-induced ventricular arrhythmias by modulating connexin43
in rats. Arch Med Res. 39:647–654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ando M, Katare RG, Kakinuma Y, Zhang D,
Yamasaki F, Muramoto K, et al: Efferent vagal nerve stimulation
protects heart against ischemia-induced arrhythmias by preserving
connexin43 protein. Circulation. 112:164–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hao JL, Suzuki K, Lu Y, Hirano S, Fukuda
K, Kumagai N, Kimura K and Nishida T: Inhibition of gap
junction-mediated intercellular communication by TNF-alpha in
cultured human corneal fibroblasts. Invest Ophthalmol Vis Sci.
46:1195–1200. 2005. View Article : Google Scholar : PubMed/NCBI
|