Introduction
Hepatitis B virus (HBV) infection can cause severe
liver diseases, including chronic hepatitis and hepatocellular
carcinoma (HCC) (1). HBV infection
remains a major health problem, with 2 billion people infected
worldwide, of which 400 million are chronically infected (1). However, the complex mechanism by
which HBV infection leads to the development of HCC remains
unclear.
It has previously been reported that the HBV X
protein (HBx) has a crucial role in hepatocarcinogenesis (2). HBx is a multifunctional protein that
activates numerous viral and cellular genes, modulates cellular
signal transduction pathways, and regulates cell proliferation and
apoptosis (3). Previous studies
have demonstrated that HBx regulates viral gene expression through
the transactivation of HBV enhancers, and may also mediate the
expression of genes in infected cells in order to facilitate
tumorigenesis (4–7).
DNA hypermethylation is a mechanism that may disrupt
the normal function of tumor suppressor genes. Aberrant methylation
of normally unmethylated CpG islands has been reported as a
relatively frequent event in immortalized and transformed cells
(4–7).
CD82 is a recently discovered tumor metastasis
suppressor gene, the expression level of which has been shown to be
highly associated with deterioration, invasion, and metastasis of
numerous human epithelial cells (7,8). The
association between CD82 expression in HCC and HBV infection has
not yet been reported. In the present study, the mRNA and protein
expression levels of CD82 in HCC tissues were determined, and the
association between CD82 and HBV was analyzed using in vitro
assays.
Materials and methods
Patients and specimens
For the present study a total of 27 patients with
HCC, admitted to Zhongnan Hospital of Wuhan University (Wuhan,
China) between January 2010 and December 2012, were retrospectively
studied. HCC was defined as liver cancer using both pathological
and clinical data. All of the patients tested positive for
hepatitis B surface antigen for >6 months. HCC tissues and
adjacent non-tumor tissues were obtained through a surgical
procedure. Written informed consent was obtained from each patient.
Ethical approval was granted by the Medical Ethical Committee of
the Zhongnan Hospital, Wuhan University (Wuhan, China) and
conducted according to the principles expressed in the Declaration
of Helsinki.
Plasmid construction
To construct the HBV plasmid pCMV-HBV-1.3, a
terminally redundant (1.3 × copy), replication-competent HBV genome
(subtype adw; nucleotides 957–1952; GenBank accession number:
AF100309) was inserted into the pUC19 (Promega Corporation,
Madison, WI, USA), as described by previous methods (9). The recombinant plasmid pCMV-HBV
contained the wild-type HBV 1.1-mer overlength genomic sequence,
and the synthesis of the pre-genomic RNA was driven by the
cytomegalovirus (CMV) promoter. A 1870 bp promoter construct of the
CD82 gene, relative to the transcriptional initiation site (nt
-1870-0), was cloned into the pGL3 luciferase construct (Promega
Corporation) and generated from human genomic DNA by polymerase
chain reaction (PCR). The resulting construct was confirmed by DNA
sequencing of CD82. A plasmid carrying 1.3-fold length of HBV
genome (pCMV-HBV1.3) and the same plasmid with a stop codon for
amino acid 7 of HBx (pCMV-HBV1.2) were generated from pCMV-HBV1.3,
which was obtained from Dr. Robert Schneider (New York University
Medical Center, New York, USA). To study the role of HBx in the
hypermethylation function, the effects of pCMV-HBV1.3 and
pCMV-HBV1.2 on the expression of CD82, were compared. There are
seven proteins in the HBV genome, including HBX, Hbe, HBs, LHBs,
MHBs, HBP and HBc. The pCMV-HBX, Hbe, HBs, LHVs, MHBs, HBP and HBc
plasmids were constructed using the pCMV-tag2B vector. To evaluate
the predominant protein involved in the regulation of CD82, these
plasmids were each transfected and the expression of CD82 was
subsequently analyzed. The plasmid pCMV-pblue and the promoter
pGL3-flag-2b were constructed and were used as controls.
Cell culture
HepG2 hepatoma cell lines were obtained from the
Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and
were characterized by mycoplasma detection, isozyme detection, DNA
fingerprinting, and cell-vitality detection. The cells were
cultured in the recommended media supplemented with 10% (vol/vol)
fetal bovine serum (Gibco-BRL, Carlsbad, CA, USA), 100 U/ml
penicillin (Invitrogen Life Technologies, Carlsbad, CA, USA), and
100 μg/ml streptomycin (Invitrogen Life Technologies) at 37°C in 5%
CO2.
Transfection
All transfections were performed using
Lipofectamine® 2000 reagent (Invitrogen Life
Technologies) according to the manufacturer’s instructions. HepG2
cells were co-transfected with different concentrations of the
pCMV-HBV1.3 plasmid and the promoter pGL3-CD82 (pHBV-1.3 group).
The relative mRNA and protein expression levels of CD82 were
detected 0, 12, 24, and 48 h post-transfection. HepG2 cells were
co-transfected with either the pCMV-HBx, pCMV-Hbe, pCMV-HBs,
pCMV-LHBs, pCMV-MHBs, pCMV-HBP or pCMV-HBc plasmid and the promoter
pGL3-CD82. As a control, HepG2 cells were co-transfected with the
plasmid pCMV-pblue and the corresponding promoter pGL3-flag-2b.
Furthermore, HepG2 cells were transiently co-transfected with
pCMV-HBV-1.2 and the promoter pGL3-CD82 (pHBV-1.2 group). The
luciferase activities, and the relative mRNA and protein expression
levels of CD82 were detected 48 h post-transfection.
Reporter assays
For the luciferase assay, HepG2 cells were
co-transfected with the plasmid and the promoter. The cell lysates
were prepared, and the luciferase activity was measured using a
luciferase assay system (Promega Corporation), 48 h
post-transfection. All transfections were performed using
Lipofectamine 2000 reagent, according to the manufacturer’s
instructions. The cell lysates (10 μl) and luciferase assay
substrates (50 μl) were mixed, and the fluorescence intensity was
detected using a luminometer (Promega Corporation). A
Renilla lucierase reporter vector, pRL-TK, was used as an
internal control. the luciferase activity was measured in each
sample 48 h after transfection using the dual-luciferase reporter
assay system (Promega Corporation) and Renilla luciferase
activities were determined as internal controls for transfection
efficiency. The assays were performed in triplicate and expressed
as the means ± standard error of the mean relative to the vector
control, which was set as 100%. All of the transfection experiments
were performed at least three times.
Reverse transcription quantitative PCR
(qPCR)
For the analysis of mRNA levels, total RNA was
extracted using TRIzol® reagent (Invitrogen Life
Technologies) according to the manufacturer’s instructions.
Quantification of total RNA was performed using a Nanodrop™
spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) at
260 and 280 nm. cDNA was synthesized using a cDNA Synthesis kit
(Toyobo, Osaka, Japan). Amplification was performed using the iQ™5
Quantitative PCR system (Bio-Rad, Hercules, CA, USA) with
SYBR® Green Master Mix (Toyobo). GAPDH was used for
normalization of the relative expression levels. The sequence of
the primers, synthesized by Invitrogen Life Technologies, used were
as follows: CD82 forward, 5′-AGGATGCCTGGGACTACGTG-3′, and reverse,
5′-GCTCAGCGTTGTCTGTCCAGT-3′; GAPDH forward,
5′-TCGTGCGTGACATTAGGAG-3′, and reverse, 5′-GTCAGGCAGCTCGTAGCTCT-3′.
The PCR conditions were set as follow: 95°C for 30 sec, 55°C for 30
sec, and 72°C for 1 min, for 40 cycles. The cycle threshold (Ct)
indicated the fractional cycle number at which the PCR product was
first detected above a fixed threshold. Relative mRNA levels were
determined using the 2−ΔΔCt method.
Western blot analysis
For the detection of protein expression levels,
cytoplasmic protein extracts were prepared from the HCC and
adjacent non-tumor tissues of the HCC patients. Tissue samples were
homogenized in a WCE buffer, which contained 26 mM HEPES (pH 7.7),
0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton
X-100, 0.5 mM dithiothritol, 20 mM glycerophosphate, 0.1 mM
Na3VO4, 2 g/ml leupeptin, 2 g/ml aprotinin, 1
mM pehnylmethylsulfonyl fluoride and a protease inhibitor cocktail
(Boehringer, Mannheim, Germany). The tissue suspension was rotated
at 4°C for 10 min and the supernatants were collected. The protein
concentration of each sample was detected by Bradford assay
(Bio-Rad Laboratories, Hercules, CA, USA). Protein (100 μg) from
each sample was separated by SDS-PAGE (4% stacking and 10%
separating gels) followed by an overnight transfer onto
polyvinylidene fluoride membranes (Millipore, Boston, MA, USA). The
membranes were then blocked for 1 h with phosphate-buffered saline
containing 0.05% Tween® 20 and 5% nonfat dry milk,
followed by an overnight incubation at 4°C with polyclonal rabbit
anti-human CD82 antibody (1:2,000 dilution; Cell Signaling
Technology, Inc., Danvers, MA, USA). The bound antibodies were
revealed using a peroxidase-labeled secondary antibody (1:5,000
dilution; Millipore) and visualized with enhanced chemiluminescence
detection reagents. The intensity of each band was quantified using
MCID Elite Software (InterFocus Imaging Ltd., Linton, UK).
Methylation assay of the CD82 gene
promoter region
To determine the effects of hypermethylation of a
CpG island within the CD82 promoter, HepG2 cells were transiently
co-transfected with the plasmid pCMV-HBV1.3 and the promoter CD82.
Bisulfite conversion of genomic DNA was performed using an EpiTect
Bisulfite kit (Qiagen, Basel, Switzerland), according to the
manufacturer’s instructions. Primers were designed for the CD82
promoter, to cover the regions containing the CpG islands. The
selected amplicon was located in the core regulatory regions of the
promoter, which covered the two glucocorticoid response elements.
The primers were designed using MethPrimer software (10). For PCR amplification, a T7-promoter
tag sequence was added to the reverse primer, and a 10-mer tag
sequence was added to the forward primer to balance the PCR primer
length. The following PCR conditions were used for the
amplification of the bisulfite-treated genomic DNA: one cycle, 94°C
for 4 min; 45 cycles, 94°C for 20 sec; 56°C for 30 sec; 72°C for 1
min; and one cycle, 74°C for 3 min. Unincorporated dinucleotide
triphosphates were removed by shrimp alkaline phosphatase (Sequenom
Inc., San Diego, CA, USA) treatment. Approximately 2 μl of the PCR
product was used as the template for the transcription reaction,
using T7 Polymerase Buffer and 20 U of T7 R&DNA™ polymerase
(Epicentre, Madison, WI, USA). In the same step, RNase A (Sequenom
Inc.) was added to cleave the in vitro transcripts
(T-cleavage assay). The samples were then diluted with
H2O to a final volume of 7 μl and incubated at 37°C for
3 h. Following the incubation, a further 20 μl of double-distilled
H2O was added to each sample. Phosphate backbone
conditioning was then performed through the addition of 6 mg of
Clean Resin (Sequenom Inc.) prior to matrix-assisted laser
desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry
analysis. A total of 12 nl of the RNase A-treated product was
robotically dispensed onto a silicon matrix of preloaded chips
(SpectroCHIP®; Sequenom Inc.), and the mass spectra were
collected using a MassARRAY® Compact MALDI-TOF (Sequenom
Inc.). The methylation ratios of the spectra were generated using
EpiTYPER software version 1.0 (Sequenom Inc.).
Statistical analyses
The data are presented as the means ± standard
deviation. Student’s t-tests were applied for the comparisons
between the groups; a P<0.05 was considered to indicate a
statistically significant difference. All statistical analyses were
performed using the statistical software SPSS version 17.0 for
Windows (SPSS Inc., Chicago, IL, USA).
Results
Expression levels of CD82 in the HCC
tissues and adjacent non-tumor tissues
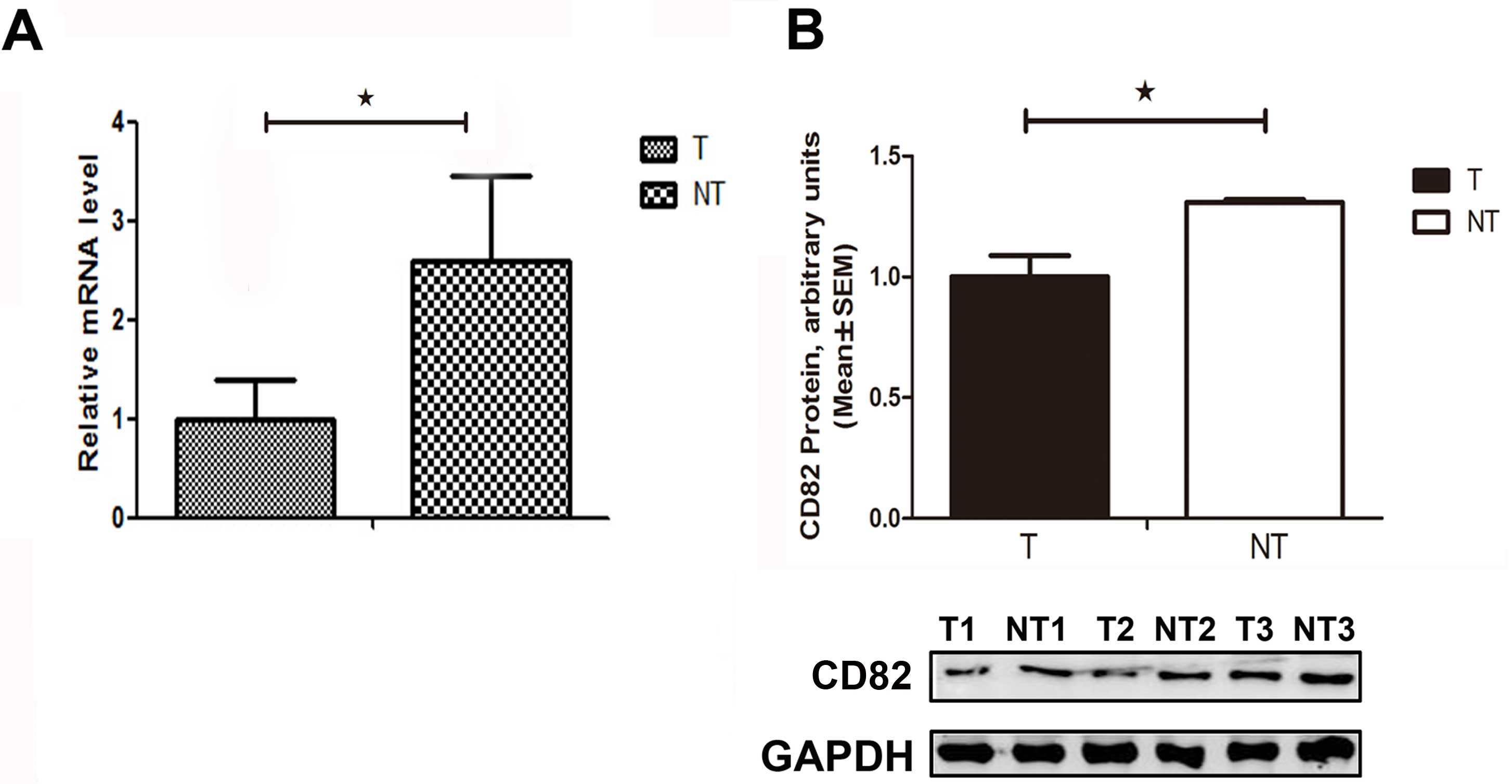
The relative mRNA expression level of CD82 was
significantly decreased by 46% (P=0.029), as compared with that in
the adjacent non-tumor tissues (Fig.
1A). The relative CD82 protein expression level was
significantly decreased in the HCC tissues, as compared with that
in the adjacent non-tumor tissues (Fig. 1B). These results suggest that CD82
expression was suppressed in HCC tissues, and CD82 gene deletion
may be an ideal target for gene therapy in the treatment of
HCC.
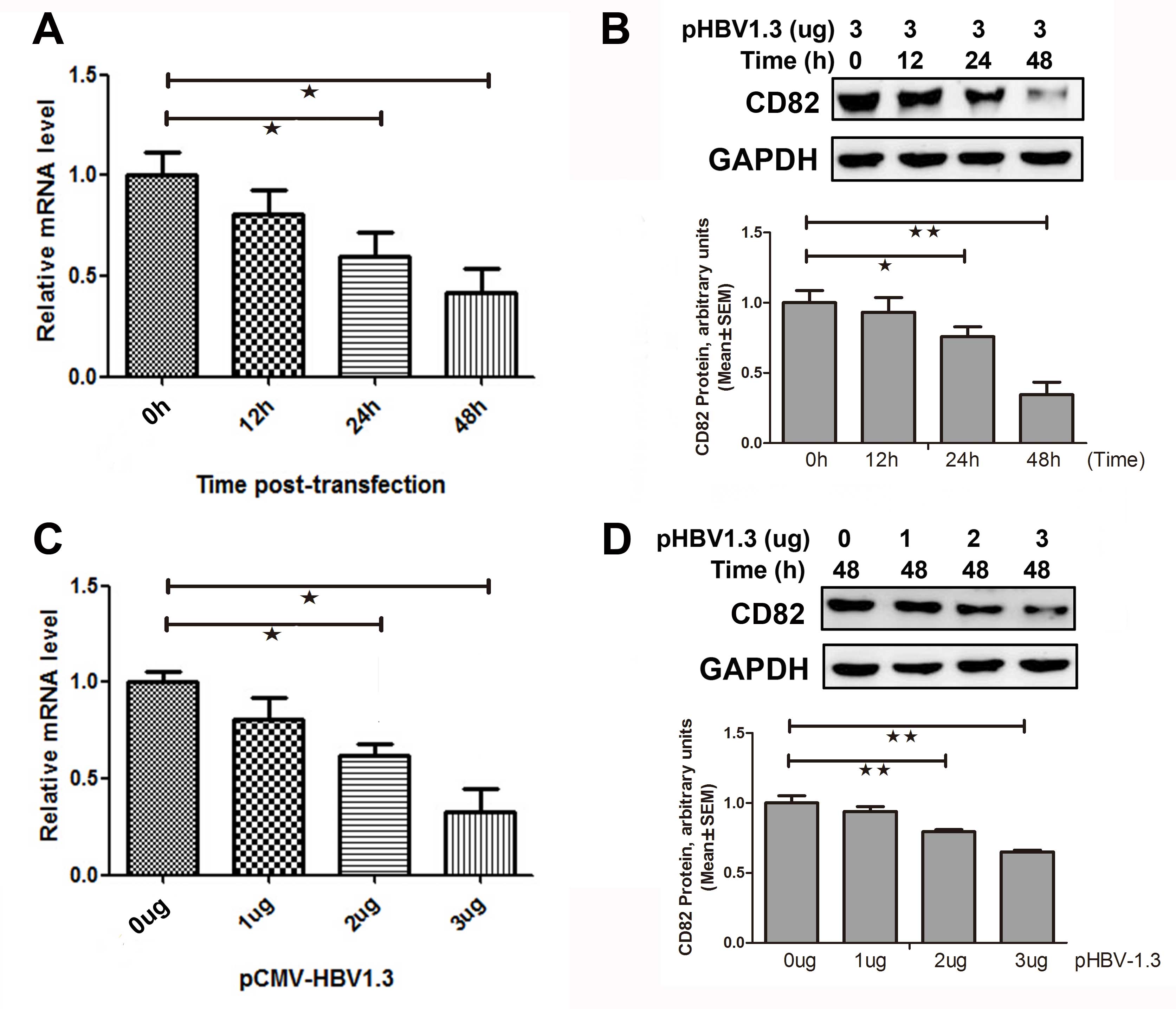
HBV inhibits CD82 mRNA and protein
expressions in vitro
HepG2 cells were transiently co-transfected with 3
μg pCMV-HBV-1.3 plasmid The relative mRNA and protein expression
levels of CD82 were determined 0, 12, 24, and 48 h
post-transfection. As shown in Fig.
2A, the mRNA expression levels of CD82 were significantly
decreased by 32 (P=0.041) and 55% (P=0.041) 24 and 48 h
post-transfection respectively, as compared to 0 h
post-transfection. The CD82 protein expression was also
significantly decreased 24 h post-transfection (0.76±0.07 vs.
1.00±0.009, P=0.014) and 48 h post-transfection (0.34±0.09 vs.
1.00±0.09, P=0.001), as compared to 0 h post-transfection (Fig. 2B).
HepG2 cells were transiently co-transfected with
various concentrations of the plasmid pCMV-HBV1.3, and the relative
mRNA and protein expression levels of CD82 were determined 48 h
post-transfection. CD82 mRNA expression was significantly decreased
by 39 (P=0.031) and 52% (P=0.015) at the concentrations of 2 and 3
μg pCMV-HBV1.3 respectively, as compared to 0 μg pCMV-HBV1.3
(Fig. 2C). As shown in Fig. 2D, the protein expression levels of
CD82 were significantly decreased when treated with 2 μg
pCMV-HBV1.3 (0.79±0.02 vs. 1.00±0.05, P=0.008) or 3 μg pCMV-HBV1.3
(0.65±0.01 vs. 1.00±0.05, P=0.001), as compared to 0 μg
pCMV-HBV1.3. These results indicate that HBV inhibited the mRNA and
protein expression levels of CD82 in vitro.
Effects of HBx on CD82 promoter activity,
mRNA and protein expression
HepG2 cells were transiently co-transfected with the
following plasmids: pCMV-HBx, pCMV-Hbe, pCMV-HBs, pCMV-LHBs,
pCMV-MHBs, pCMV-HBP, pCMV-HBc and the promoter pGL3-CD82. The
luciferase activity of CD82 was detected 48 h post-transfection.
The plasmid pCMV-pblue and the corresponding promoter pGL3-flag-2b
was constructed and used as a control. As shown in Fig. 3A, the luciferase activity of CD82
was significantly decreased by 60% (P=0.005) following
co-transfection with the plasmid pCMV-HBx and the promoter
pGL3-CD82 (HBx group), as compared with the luciferase activity of
the control group. HepG2 cells were transiently transfected with
the following plasmids: pCMV-HBx, pCMV-Hbe, pCMV-HBs, pCMV-LHBs,
pCMV-MHBs, pCMV-HBP and pCMV-HBc. The plasmid pCMV-pblue was used
as a control. The mRNA and protein expression levels of CD82 were
detected 48 h post-transfection. The CD82 mRNA expression level in
the HBx group was decreased by 62% (P=0.024), as compared with the
control group (Fig. 3B). As shown
in Fig. 3C, the protein expression
level of CD82 in the HBx group was also significantly decreased, as
compared with the control group.
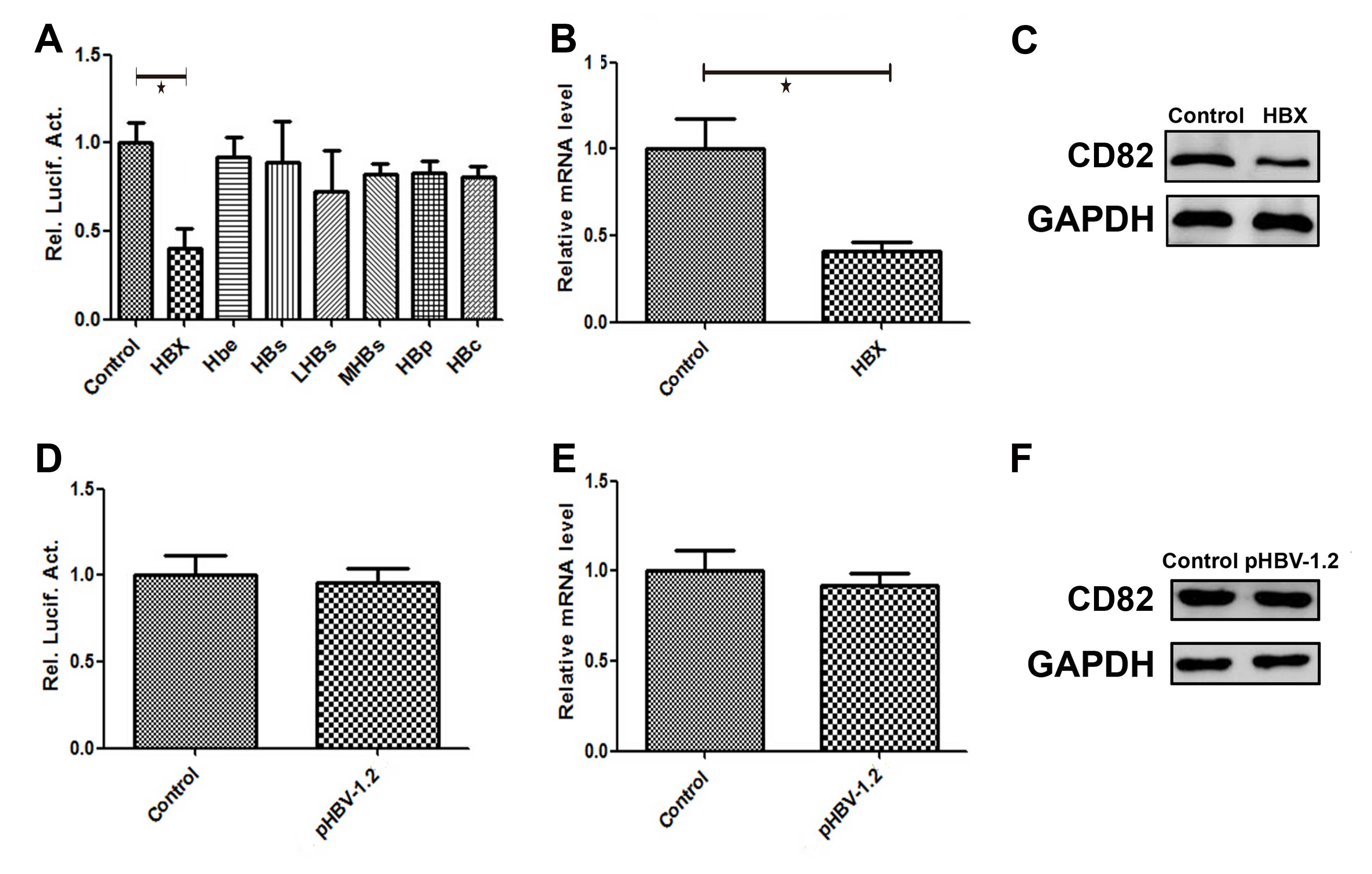 | Figure 3Effects of hepatitis B virus X protein
(HBx) on CD82 promoter activity, mRNA and protein expression. HepG2
hepatoma cells were transiently co-transfected with the plasmids
pCMV-HBx, pCMV-Hbe, pCMV-HBs, pCMV-LHBs, pCMV-MHBs, pCMV-HBP,
pCMV-HBc and the promoter pGL3-CD82. (A–C) The luciferase activity,
and the relative mRNA and protein expression levels of CD82 were
significantly decreased in the HBx group, as compared with the
control group. HepG2 cells were transiently co-transfected with the
plasmids pCMV-HBV-1.2 and the promoter pGL3-CD82 (pHBV-1.2 group).
(D–F) The luciferase activity, and relative mRNA and protein
expression levels of CD82 were no different as compared with the
control group. |
HepG2 cells were transiently co-transfected with the
plasmid pCMV-HBV-1.2 and the promoter pGL3-CD82 (pHBV-1.2 group).
The luciferase activity, and the relative mRNA and protein
expression levels of CD82 were detected 48 h post-transfection. The
plasmid pCMV-pblue and the corresponding promoter pGL3-flag-2b was
constructed and used as a control. As shown in Fig. 3D, there was no difference in the
luciferase activity of CD82 in the pHBV-1.2 group, compared to the
control group. The mRNA and protein expression levels of CD82 were
detected 48 h post-transfection. tHe plasmid pCMV-pblue was used as
a control. As shown in Fig. 3E and
F, there was no difference in the mRNA and protein expressions
of CD82 in the pHBV-1.2 group, compared to the control group.
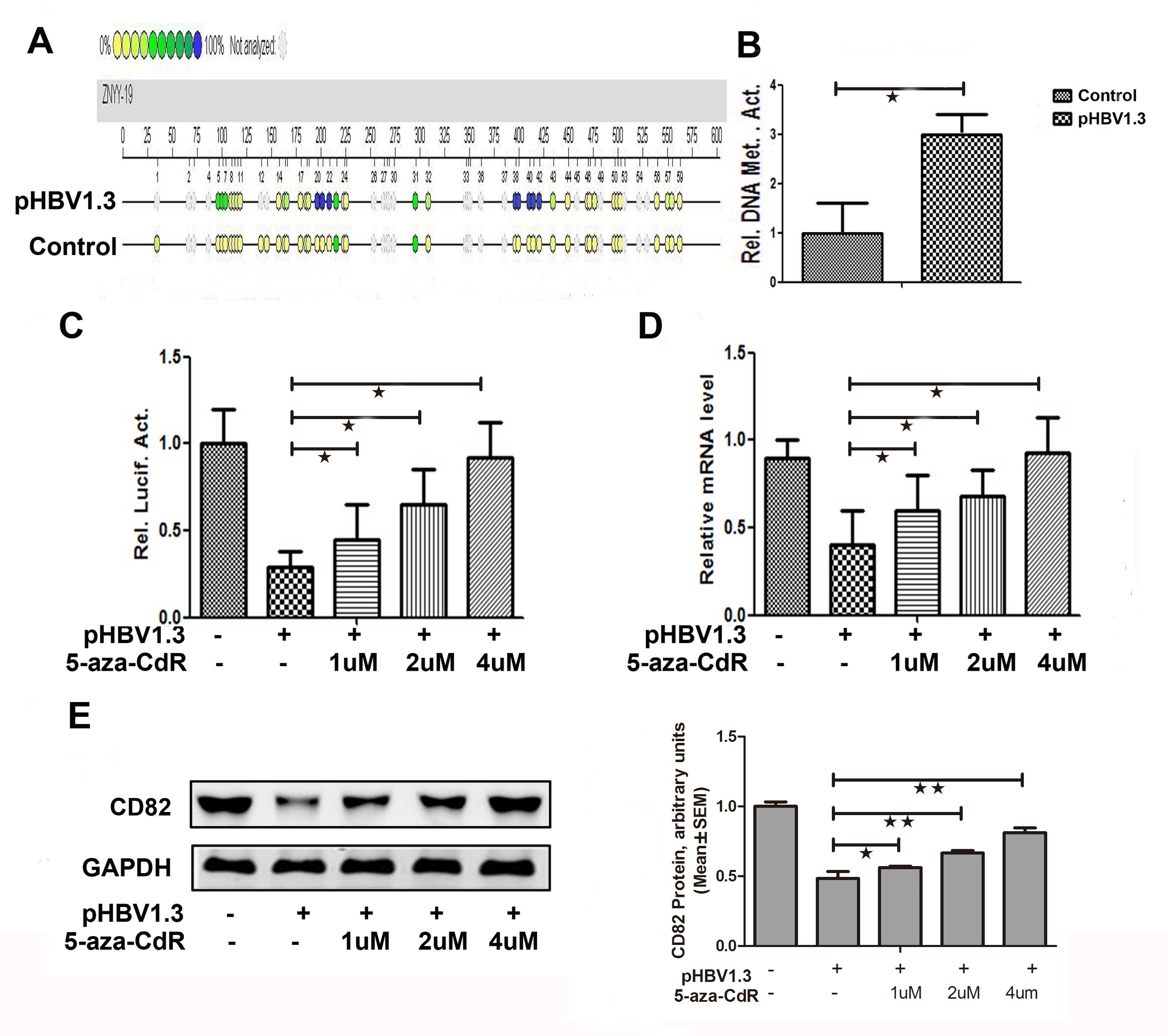
Effects of HBV on CD82 expression by
inducing CD82 promoter methylation
The overall methylation level of the CD82 promoter
in the pHBV-1.3 group was significantly increased by 3-fold
(P=0.018), as compared with that in the control group (Fig. 4A and B). As shown in Fig. 4C, the promoter activities of CD82
was increased by 1.53 (P=0.032), 2.17 (P=0.019) and 3.14-fold
(P=0.027) when treated with 1, 2 and 4 μM of 5-aza-CdR
respectively, in the pHBV-1.3 group. The CD82 mRNA expression
levels were increased by 1.36 (P=0.033), 1.59 (P=0.032) and
2.27-fold (P=0.024) when treated with 1, 2 and 4 μM of 5-aza-CdR
respectively, in the pHBV-1.3 group (Fig. 4D). The protein expression levels of
CD82 were also increased in the pHBV-1.3 group, when treated with 1
μM (0.56±0.01 vs. 1.00±0.03, P−=0.040), 2 μM (0.66±0.02 vs.
1.00±0.03, P=0.002), or 4 μM (0.81±0.03 vs. 1.00±0.03, p<0.001)
of 5-aza-CdR (Fig. 4E). These
results indicate that the inhibitor of methylation 5-aza-CdR could
reverse the effects of HBV on CD82. Therefore, it may be
hypothesized that HBV may affect the expression of CD82 by inducing
CD82 promoter methylation.
Discussion
CD82 is a tumor metastasis suppressor gene and one
of four transmembrane glycoprotein superfamily members. The CD82
gene is located on the human chromosome 11p11.2, and it encodes a
267 amino acid protein molecule, the molecular weight of which is
29 kDa (11). The downregulation
of CD82 may be an important factor in the development of liver
cancer, however the exact mechanisms by which this occurs remain to
be elucidated. Previous studies have shown that CD82 inhibits the
metastasis of a vast majority of tumors, and is associated with
tumor invasiveness (11). CD82
expression has also been shown to be highly associated with
deterioration, invasion, and metastasis of numerous human
epithelial cells (10,11). However, the association between the
CD82 gene and HBV in HCC has not yet been determined. Whether HBV
can regulate the development of liver cancer through influencing
CD82 gene expression is still unclear (12,13).
DNA methylation is a chemical modification method of
the eukaryotic genome. DNA methylation is the process by which
methyl groups from S-adenosylmethionine are transferred to the 5′
cytosine molecule of a carbon ring, resulting in the formation of a
5-methylcytosine. Methylation is catalyzed by the DNA
methyltransferase enzyme. The repeated sequences of certain genes
in healthy cells have high methylation levels which prevent
reactivation of transcription factors, resulting in genomic
stability (14–16). If the normal methylation of somatic
cells changes, known as whole genome hypomethylation, it may lead
to uncontrolled cell growth. Changing site-specific methylation,
for example tumor suppressor gene promoter CpG island
hypermethylation, may lead to tumorigenesis. This change is an
epigenetic characteristic of tumor cells, and is present in almost
all types of tumors (17–19). There are numerous key cancer gene
promoter regions which are involved in DNA repair, cell cycle
regulation, resistance formation and tumor invasion, metastasis and
angiogenesis, and the presence of high methylation status may be a
tumor characteristic (17).
Previous studies have determined that hypermethylation of the
promoter region leads to the inactivation of some tumor suppressor
genes, which is a common feature of human tumors (18). Furthermore, CD82 hypermethylation
may lead to the inactivation of tumor suppressor genes. The
localized hypermethylation of tumor cells generally occurs in the
promoter region of the gene, which may result in the silenced
expression of the gene (19). A
previous study found that in the development of liver cancer, some
viruses could cause tumor suppressor gene promoter
hypermethylation, resulting in the transcriptional interference of
tumor suppressor gene expression, abnormal cell proliferation and
cancer development (20).
Considering the close association between DNA methylation and human
cancer development, especially due to CpG island methylation of
tumor suppressor genes, DNA methylation and epigenetics have
recently become an important area of study.
Liver cancer is a common primary tumor, with
characteristics including recurrence, metastasis, high mortality
and poor prognosis (21). Previous
studies have reported that 80% of liver cancer cases are
accompanied by HBV infection (22,23).
It is important for future research to identify specific biomarkers
for the early diagnosis of liver cancer, as well as looking for a
new target for treatment. The present study demonstrated that
aberrant epigentic modifications in the promoter of CD82 were
induced by HBx. Hypermethylation of the promoter of CD82 lead to
the reduction of expression, which may subsequently participate in
the prgression of HCC. The results indicated that induciton of CD82
expression by treatment with the methyl enzyme inhibitor 5-aza-CdR,
could be a potential treatment for HBV-induced HCC. In conclusion,
the present study provided a theoretical basis for the clinical
treatment of HBV-induced cancer.
Acknowledgements
This work was supported by grants from the National
Natural Science Foundation of China (nos. 30872491/C160402,
81372552 and 81172349/H1617). The authors would like to thank all
the gastroenterologists and investigators in the Research Center of
Digestive Diseases.
References
|
1
|
Benhenda S, Cougot D, Buendia MA and
Neuveut C: Hepatitis B virus X protein molecular functions and its
role in virus life cycle and pathogenesis. Adv Cancer Res.
103:75–109. 2009.PubMed/NCBI
|
|
2
|
Feitelson MA and Lee J: Hepatitis B virus
integration, fragile sites, and hepatocarcinogenesis. Cancer Lett.
252:157–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chan DW and Ng IO: Knock-down of hepatitis
B virus X protein reduces the tumorigenicity of hepatocellular
carcinoma cells. J Pathol. 208:372–380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng AS, Wong N, Tse AM, Chan KY, Chan
KK, Sung JJ and Chan HL: RNA interference targeting HBx suppresses
tumor growth and enhances cisplatin chemosensitivity in human
hepatocellular carcinoma. Cancer Lett. 253:43–52. 2007. View Article : Google Scholar
|
|
5
|
Zhang X, Zhang H and Ye L: Effects of
hepatitis B virus X protein on the development of liver cancer. J
Lab Clin Med. 147:58–66. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moon EJ, Jeong CH, Jeong JW, Kim KR, Yu
DY, Murakami S, Kim CW and Kim KW: Hepatitis B virus X protein
induces angiogenesis by stabilizing hypoxia-inducible
factor-1alpha. FASEB J. 18:382–384. 2004.PubMed/NCBI
|
|
7
|
Robinson WS: Molecular events in the
pathogenesis of hepadnavirus-associated hepatocellular carcinoma.
Annu Rev Med. 45:297–323. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nephew KP and Huang TH: Epigenetic gene
silencing in cancer initiation and progression. Cancer Lett.
190:125–133. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, Lin S, Chen Q, Peng L, Zhai J, Liu Y
and Yuan Z: Inhibition of hepatitis B virus replication by MyD88
involves accelerated degradation of pregenomic RNA and nuclear
retention of pre-S/S RNAs. J Virol. 84:6387–6399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li LC and Dahiya R: MethPrimer: designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong JT, Lamb PW, Rinker-Schaeffer CW,
Vukanovic J, Ichikawa T, Isaacs JT and Barrett JC: CD82, a
metastasis suppressor gene for prostate cancer on human chromosome
11p11.2. Science. 268:884–886. 1995. View Article : Google Scholar
|
|
12
|
Miranti CK: Controlling cell surface
dynamics and signaling: how CD82/CD82 suppresses metastasis. Cell
Signal. 21:196–211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ai X, Zhang X, Wu Z, Ma X, Ju Z, Wang B
and Shi T: Expression of CD82/CD82 and MRP-1/CD9 in transitional
cell carcinoma of bladder. J Huazhong Univ Sci Technolog Med Sci.
27:79–82. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nelson WG, Yegnasubramanian S, Agoston AT,
Bastian PJ, Lee BH, Nakayama M and De Marzo AM: Abnormal DNA
methylation, epigenetics, and prostate cancer. Front Biosci.
12:4254–4266. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dalmay T and Edwards DR: MicroRNAs and the
hallmarks of cancer. Oncogene. 25:6170–6175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kent OA and Mendell JT: A small piece in
the cancer puzzle: microRNAs as tumor suppressors and oncogenes.
Oncogene. 25:6188–6196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu J: DNA methylation and hepatocellular
carcinoma. J Hepatobiliary Pancreat Surg. 13:265–73. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang B, Guo M, Herman JG and Clark DP:
Aberrant promoter methylation profiles of tumor suppressor genes in
hepatocellular carcinoma. Am J Pathol. 163:1101–1107. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ and
Kang GH: Aberrant CpG island hypermethylation along multistep
hepatocarcinogenesis. Am J Pathol. 163:1371–1378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pogribny IP and Rusyn I: Role of
epigenetic aberrations in the development and progression of human
hepatocellular carcinoma. Cancer Lett. 342:223–230. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shiraha H, Yamamoto K and Namba M: Human
hepatocyte carcinogenesis (review). Int J Oncol. 42:1133–1138.
2013.PubMed/NCBI
|
|
22
|
Li LH, He J, Hua D, Guo ZJ and Gao Q:
Lentivirus-mediated inhibition of Med19 suppresses growth of breast
cancer cells in vitro. Cancer Chemother Pharmacol. 68:207–215.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perz JF, Armstrong GL, Farrington LA,
Hutin YJ and Bell BP: The contributions of hepatitis B virus and
hepatitis C virus infections to cirrhosis and primary liver cancer
worldwide. J Hepatol. 45:529–538. 2006. View Article : Google Scholar : PubMed/NCBI
|