Introduction
Previous studies have demonstrated that physical
exercise had neuroprotective effects in animals following ischemic
stroke, including enhanced survival, decreased neurological
deficits, reduced blood-brain barrier (BBB) dysfunction and
increased neurovascular integrity (1–5).
However, the mechanism underlying the neuroprotective effect of
exercise have remained to be elucidated.
Excessive release of glutamate has been confirmed to
have a key role in the process of brain damage following ischemic
stroke and is involved in the majority of ischemic
stroke-associated brain injuries (6). A previous study reported that three
weeks of pre-ischemic treadmill training may ameliorate the
increased release of glutamate (7)
and decrease messenger (m)RNA levels of metabotropic glutamate
receptor 5 and N-methyl-D-aspartate receptor subunit type 2B
(8), which resulted in alleviation
of the toxic effects of excessive glutamate.
Glutamate transporters also have an important role
in glutamate re-uptake, which contribute to the pathological
mechanisms of glutamate-induced toxic injury. Five excitatory amino
acid transporters (EAATs) have been identified in the mammalian
CNS, of which EAAT-2 was an important transporter for the
regulation of extracellular glutamate concentrations (9,10).
Therefore, the hypothesis of the present study was that
pre-ischemic exercise intervention promotes the expression of
EAAT-2 so as to reduce brain damage following ischemic stroke.
The extracellular signal-regulated kinase 1/2
(ERK1/2) pathway has been reported to have an important role in the
neuroprotective effect of exercise preconditioning (11). ERK1/2 is a key subfamily of
mitogen-activated protein kinases which regulate a broad range of
cellular activities, including the protection against cerebral
ischemia (12). Inhibition of the
ERK1/2 pathway was previously reported to decrease brain damage and
infarct volume in mice following ischemic stroke (13,14);
in addition, another study showed that following three weeks of
pre-ischemic exercise the expression of phosphorylated
(phospho)-ERK1/2 was downregulated 48 h post-reperfusion (11). However, the effect of pre-ischemic
exercise on the expression of phospho-ERK1/2 at 24 h
post-reperfusion has remained to be elucidated.
The aim of the present study was to investigate
whether three weeks of exercise preconditioning reduced
neurological deficits, infarct size as well as altered the
expression of phospho-ERK1/2 and EAAT-2.
Materials and methods
Animals
A total of 36 male Sprague-Dawley rats (weight,
200–220g; age, 2 months) were provided by the Hebei Province
Laboratory Animal Center (Hebei, China). Rats were housed under
standard conditions in a 12-h light/dark cycle, with food and water
available ad libitum. All procedures in the present study
were approved by the Animal Care and Use Committee of Hebei Medical
University (Hebei, China).
Treadmill training
Rats were divided at random into three groups as
follows (n=12 for each): Sham surgery group; middle cerebral artery
occlusion (MCAO) group; and exercise with MCAO group. Prior to
formal training, rats in the exercise with MCAO group were
subjected to running exercise for three days at a speed of 6–7
m/min for 20 min per day using a treadmill training machine
(DSPT-202 Type 5-Lane Treadmill; Litai Biotechnology Co., Ltd,
Shishi, China). Following adaptive exercise training, rats
underwent formal exercise intervention at a speed of 20 m/min for
30 min per day, six times per week. Rats in the sham surgery and
MCAO groups were enabled to run freely in their cages in the
corresponding period.
MCAO model
Following treadmill training, all rats underwent
MCAO or sham surgery. The MCAO model rats were anesthetized
(Sigma-Aldrich, Poole, UK) using an intraperitoneal injection of 4%
chloral hydrate (10 ml/kg); further doses were required if rats did
not remain anesthetized during surgery. A heating pad was used to
maintain the rats’ body temperature at 37°C. Surgical procedures
were performed as previously described by Longa et al
(15).
In brief, the left common carotid artery (CCA),
external carotid artery (ECA) and internal carotid artery (ICA)
were exposed. A suture (4-0 nylon) with a rounded
poly-L-lysine-coated tip (Beijing Sunbio Biotech Co., Ltd, Beijing,
China) was inserted into the small incision in the ECA. The suture
was then passed through CCA and ICA to the origin section of the
MCA in order to occlude this artery. Following 90 minutes of
occlusion, the filament was removed to allow for reperfusion.
Rats in the sham group underwent the same procedures
without occlusion of MCA. The physiological parameters of rats were
determined using a Blood Gas and Electrolyte System (ABL505;
Radiometer, Copenhagen, Denmark). Rats were evaluated 24 h
following reperfusion in accordance with a widely used scale as
follows: 0, no neurological symptoms; 1, unable to completely
extend the front jaw on the contralateral side; 2, rotating while
crawling and falling to the contralateral side; 3, unable to walk
without assistance; and 4, unconsciousness (15).
Determination of brain infarct
volume
Following 24 h post-reperfusion, rats were
anesthetized using chloral hydrate (10%) and then sacrificed. Brain
tissues were stored at −20°C for 10 minutes, then the whole brain
was divided evenly into six coronal sections (2 mm) from the
anterior pole to the optic chiasm in the mid-way. Brain sections
were immediately dipped into a 2% 2,3,5-triphenyltetrazolium
chloride solution (Sigma-Aldrich) at 37°C for 30 min and then fixed
in 4% paraformaldehyde buffer (Sigma, Muenchen, Germany). Following
24 h, the infarct area was calculated according to the images
captured using a digital camera (DC240; Kodak, Rochester, NY, USA)
and imaging software (Adobe Photoshop 7.0; Adobe Systems, San Jose,
CA, USA). The infarction volume of whole brain was equal to the sum
of the infarct area in the six sections. In order to reduce error
due to brain edema, the corrected formula to calculate the infarct
volume was as follows (16):
Infarct volume = contralateral hemisphere region - non-infarcted
region in the ipsilateral hemisphere; infarct percentage = (Infarct
volume/volume of the contralateral hemisphere)x100%.
Western blot analysis
Cortex brain tissue in proximity to the ischemic
area was examined. The protein of the brain tissue was extracted
using a protein extraction reagent (Pierce Biotechnology, Inc.,
Rockford, IL, USA). The extracted proteins were purified by
affinity chromatography and the concentration of extracted protein
was detected using the bicinchoninic acid assay (Beyotime Institute
of Biotechnology, Haimen, China). Equal quantities (40 μg) of
protein extract and sample buffer were mixed completely and
incubated in a 95°C-water bath for 5 min prior to loading onto 10%
polyacrylamide gels. The proteins were delivered onto a
HybondTM nylon membrane (GE Healthcare, Little Chalfont,
UK) at 350 V for 90 min with a cold pack (at 0°C). Proteins were
then stored in 5% bovine serum albumin blocking solution for 1 h at
25°C and incubated overnight at 40°C with monoclonal rabbit
anti-ERK1/2 (dilution, 1:1,000), monoclonal rabbit
anti-phospo-ERK1/2 (dilution, 1:1,000) or polyclonal rabbit
anti-EAAT-2 antibodies (dilution, 1:1,000) (Cell Signaling
Technology, Inc., Danvers, MA, USA). Protein was then incubated
with horseradish peroxidase-labeled anti-rabbit secondary
antibodies (dilution, 1:100; Hua-Mei Biotech, Beijing, China) for 1
h at room temperature with blocking buffer. The membrane was then
visualized using the enhanced chemiluminescence kit (GE Healthcare)
for 5 min and exposed to Kodak film (Kodak) for 5–30 seconds. GAPDH
or β-actin was used as a loading control.
Statistical analysis
SPSS version 15.0 (SPSS Inc, Chicago, IL, USA) was
used to perform data analysis. Neurological deficit scores and
infarct volumes between the MCAO and exercise with MCAO groups were
compared using Student’s t-test. Relative image density among the
three groups analyzed using a one-way analysis of variance was
followed by a least significant difference post-hoc test. Data are
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference
between values.
Results
Physiological variables are unchanged
between the groups
Physiological parameters were evaluated in rats from
each group (data not shown). The parameters investigated included
hydrogen ion concentration, partial pressure of carbon dioxide in
the arteries and partial pressure of oxygen in the arterial blood.
No significant differences were identified among the parameters of
the the three groups (P>0.05).
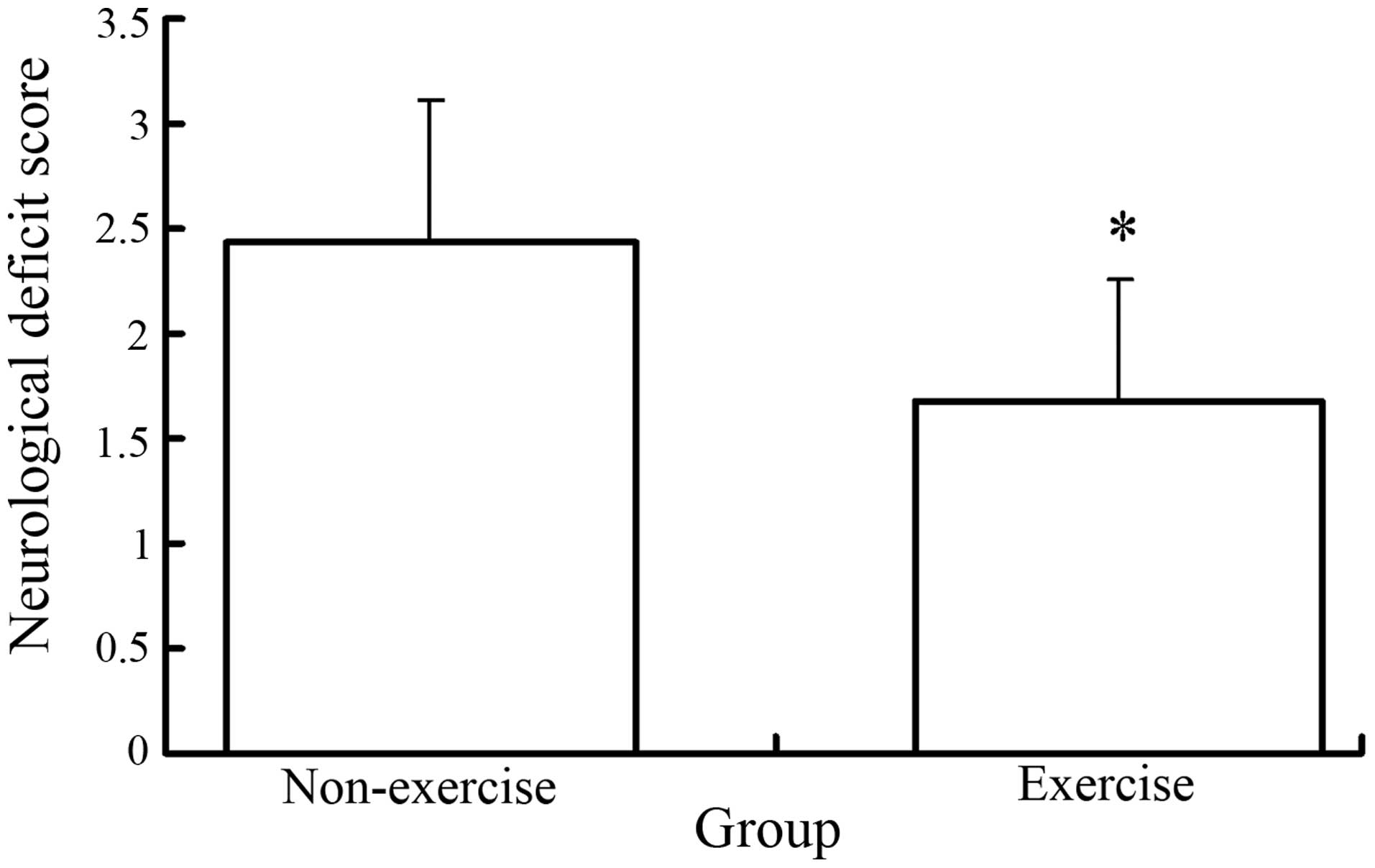
Exercise reduces post-reperfusion
neurological deficits
Neurological evaluations were performed 24 h
post-reperfusion and their scores calculated. Rats in the sham
surgery group demonstrated no neurological symptoms; however, the
neurological deficit scores for the exercise with MCAO group were
significantly lower than those of the MCAO group (P<0.05)
(Fig. 1).
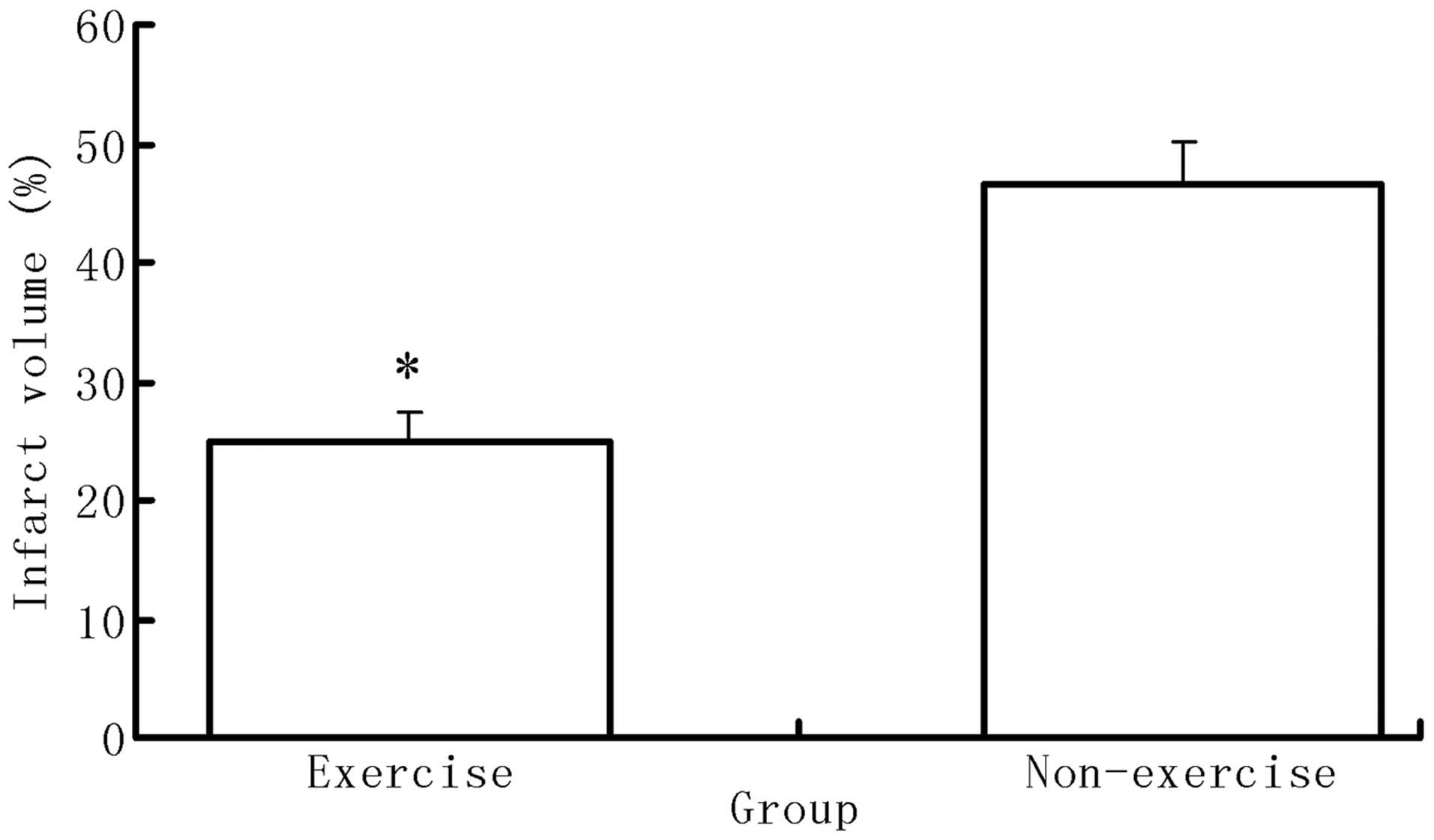
Exercise reduces infarct volume following
MCAO surgery
Following neurological evaluation, six rats from
each group were sacrificed in order to calculate the infarct
volume. No ischemic areas were identified in the brains of rats in
the sham group; however, rats of the exercise with MCAO group
showed significantly decreased volumes of infarction compared with
those of the MCAO group (P<0.05) (Fig. 2).
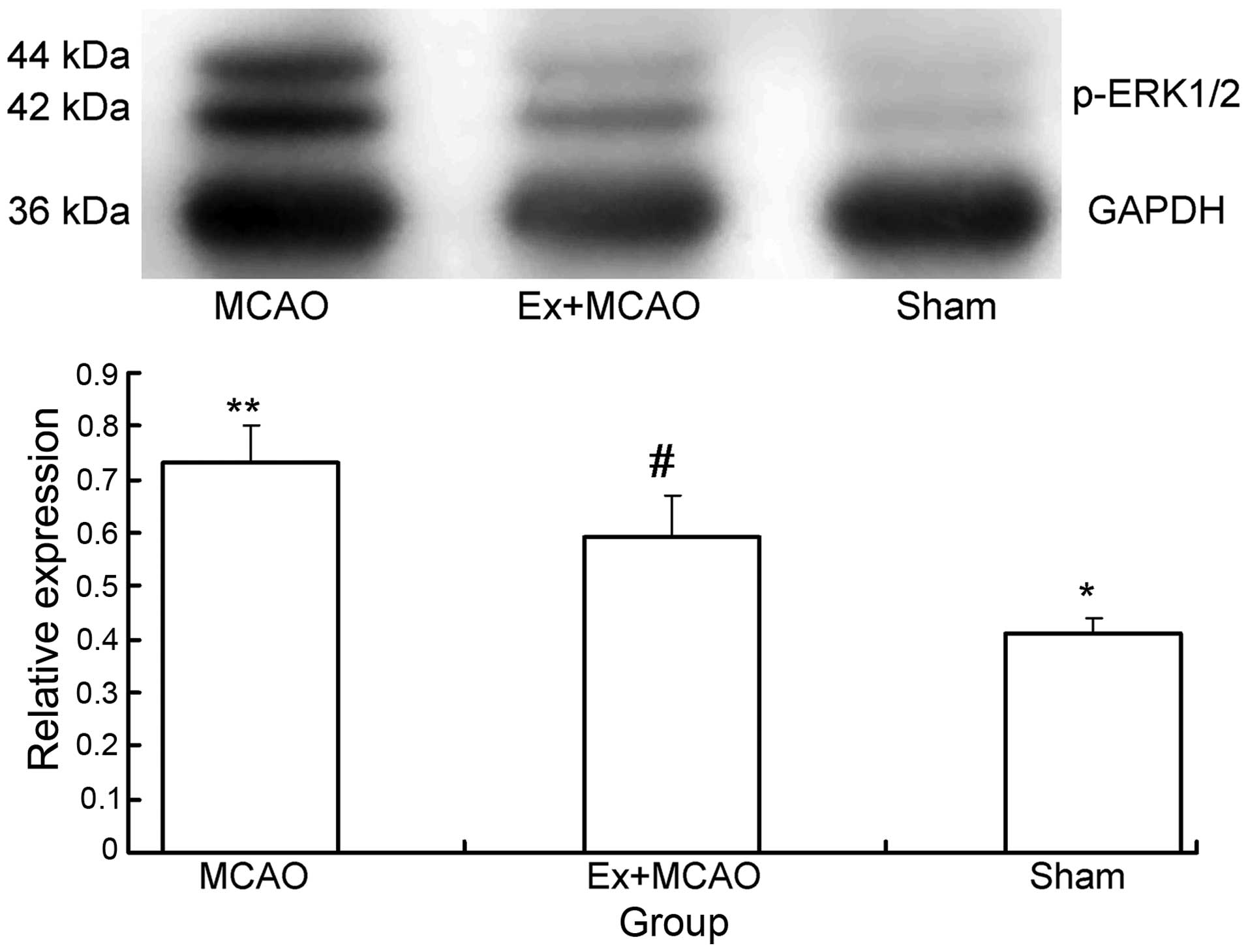
Exercise reduces phospho-ERK1/2
overexpression following ischemic brain injury
Western blot analysis was used to determine the
protein levels of ERK1/2 and phospho-ERK1/2 in rats from each
group. As shown in Figs. 3 and
4, total ERK1/2 expression was not
significantly different among the three groups; however, there were
significant differences in phospho-ERK1/2 expression levels among
the three groups. MCAO rats demonstrated increased phospho-ERK1/2
expression compared to that of the sham surgery group (P<0.05);
in addition, the pre-ischemic exercise group showed decreased
phospho-ERK1/2 compared to that of the MCAO group (P<0.05),
indicating that pre-ischemic exercise alleviated this effect.
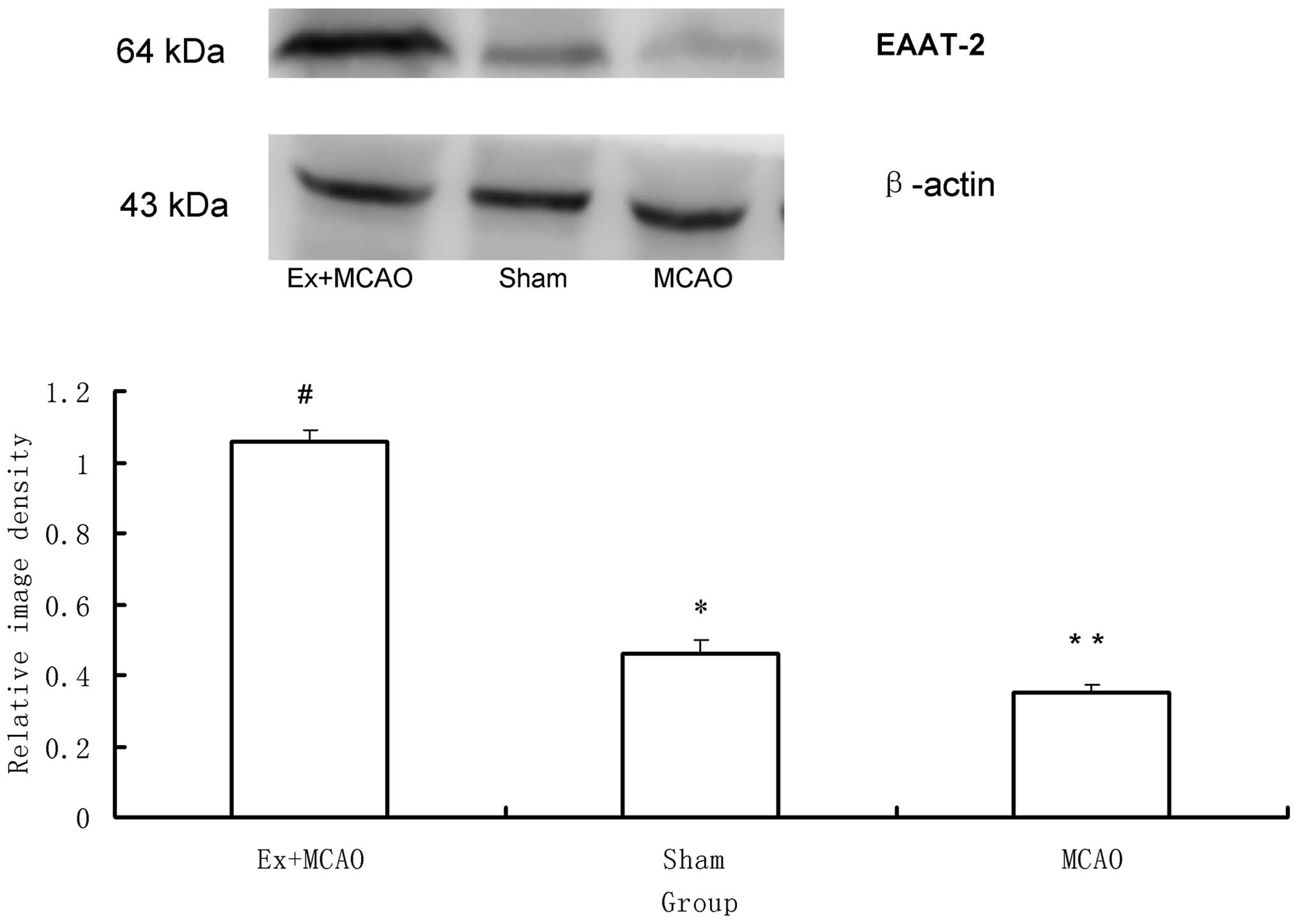
EAAT-2
As shown in Fig. 5,
western blot analysis revealed significant differences in EAAT-2
protein expression among the three groups (P<0.05). EAAT-2 was
markedly decreased in the MCAO group compared to that of the sham
surgery group and was markedly increased in the pre-ischemic
exercise group compared to that of the MCAO group (P<0.05).
Discussion
Ischemic stroke is a prevalent cause of mortality in
Western countries (17);
therefore, studies into the alleviation of brain damage following
ischemic stroke are essential. A previous study investigated a
series of intervention methods and reported that physical exercise
may be effective in alleviating brain injury following ischemic
stroke (18); however, the
mechanisms underlying the neuroprotective effects of exercise prior
to ischemic brain injury required elucidation in order to encourage
individuals with high risk factors of stroke to begin to exercise
regularly.
In the pathological process of cerebral
ischemia-reperfusion injury, neurotoxicity due to glutamate
over-release is a primary damaging factor (6). EAATs were reported to have a key role
in the uptake process of excess extracellular glutamate (9,10),
and they were found to downregulate glutamate uptake ability,
resulting in an ~65% increase of glutamate concentration following
diffuse brain injury (19). EAAT-2
is expressed in astrocytes of different brain areas, particularly
in the cerebral cortex and hippocampus (20) and was reported to be responsible
for 90% of glutamate re-uptake into the prosencephalon of rats
(21). EAAT-2 knockout mice showed
selective neuronal deterioration in the area of hippocampal CA1,
indicating the role of EAAT-2 in neuroprotection (22). The results of the present study
demonstrated that pre-ischemic exercise enhanced expression levels
of EAAT-2, therefore alleviating the neurotoxicity caused by
excessive release of glutamate.
ERK1/2 was reported to have different roles in the
pre- and post-stroke phases (11–14).
Previous studies have indicated that an inhibitor of
mitogen-activated protein kinase (MAPK)/ERK (MEK1) alleviated brain
damage of mice following ischemic stroke, indicating that the
MEK1-ERK1/2 pathway was involved in cerebral damage in the process
of cerebral ischemia (13–14). Therefore, intervention which may
decrease the overexpression of phospho-ERK1/2 following ischemic
stroke may alleviate ischemia-induced damage.
In a previous study, the promotion of phospho-ERK1/2
induced by exercise preconditioning was found to be neuroprotective
(11). Another study demonstrated
that pre-ischemic exercise decreased overexpression of
phospho-ERK1/2 at 48 hours following ischemia/reperfusion (8). The results of the present study
further confirm that exercise preconditioning alleviated the
overexpression of phospho-ERK1/2 at 24 h following
ischemia/reperfusion.
In conclusion, the results of the present study
indicated that pre-ischemic exercise exerted a neuroprotective
effect via the decreased overexpression of phospho-ERK1/2 and
increased expression of EAAT-2 following cerebral ischemia. Further
patient studies are required in order to confirm the beneficial
effect of pre-ischemic exercise in humans; however, it is
encouraged that patients with a high risk of stroke exercise
regularly.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81201512).
References
|
1
|
Stummer W, Baethmann A, Murr R, et al:
Cerebral protection against ischemia by locomotor-activity in
gerbils. Underlying mechanisms. Stroke. 26:1423–1429. 1995.
View Article : Google Scholar
|
|
2
|
Ang ET, Wong PTH, Moochhala S and Ng YK:
Neuroprotection associated with running: is it a result of
increased endogenous neurotrophic factors? Neuroscience.
118:335–345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Endres M, Gertz K, Lindauer U, et al:
Mechanisms of stroke protection by physical activity. Ann Neurol.
54:582–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li J, Luan XD, Clark JC, et al:
Neuroprotection against transient cerebral ischemia by exercise
pre-conditioning in rats. Neurol Res. 26:404–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding YH, Ding Y, Li J, et al: Exercise
pre-conditioning strengthens brain microvascular integrity in a rat
stroke model. Neurol Res. 28:184–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guyot LL, Diaz FG, O’Regan MH, McLeod S,
Park H and Phillis JW: Real-time measurement of glutamate release
from the ischemic penumbra of the rat cerebral cortex using a focal
middle cerebral artery occlusion model. Neurosci Lett. 299:37–40.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang F, Jia J, Wu Y, Hu Y and Wang Y: The
effect of treadmill training pre-exercise on glutamate receptor
expression in rats after cerebral ischemia. Int J Mol Sci.
11:2658–2669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang F, Wu Y, Jia J and Hu YS:
Pre-ischemic treadmill training induces tolerance to brain
ischemia: involvement of glutamate and ERK1/2. Molecules.
15:5246–5257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beart PM and O’Shea RD: Transporters for
L-glutamate: an update on their molecular pharmacology and
pathological involvement. Br J Pharmacol. 150:5–17. 2007.
View Article : Google Scholar
|
|
10
|
Suchak SK, Baloyianni NV, Perkinton MS,
Williams RJ, Meldrum BS and Rattray M: The ‘glial’ glutamate
transporter, EAAT2 (Glt-1) accounts for high affinity glutamate
uptake into adult rodent nerve endings. J Neurochem. 84:522–532.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liebelt B, Papapetrou P, Ali A, et al:
Exercise preconditioning reduces neuronal apoptosis in stroke by
up-regulating heat shock protein-70 (heat shock protein-72) and
extracellular signal-regulated-kinase 1/2. Neuroscience.
166:1091–1100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu ZM and Xu SC: ERK1/2 MAP kinases in
cell survival and apoptosis. IUBMB Life. 58:621–631. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alessandrini A, Namura S, Moskowitz MA and
Bonventre JV: MEK1 protein kinase inhibition protects against
damage resulting from focal cerebral ischemia. Proc Natl Acad Sci
USA. 96:12866–12869. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Namura S, Iihara K, Takami S, et al:
Intravenous administration of MEK inhibitor U0126 affords brain
protection against forebrain ischemia and focal cerebral ischemia.
Proc Natl Acad Sci USA. 98:11569–11574. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ding YH, Ding Y, Li J, Bessert DA and
Rafols JA: Exercise pre-conditioning strengthens brain
microvascular integrity in a rat stroke model. Neurol Res.
28:184–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
National Center for Health Statistics.
Health, United States, 2010: With Special Feature on Death and
Dying. Hyattsville (MD, USA): 2011
|
|
18
|
Krarup LH, Truelsen T, Gluud C, et al:
ExStroke Pilot Trial Group: Prestroke physical activity is
associated with severity and long-term outcome from first-ever
stroke. Neurology. 71:1313–1318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hinzman JM, Thomas TC, Quintero JE,
Gerhardt GA and Lifshit J: Disruptions in the regulation of
extracellular glutamate by neurons and glia in the rat striatum two
days after diffuse brain injury. J Neurotrauma. 29:1197–1208. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanai Y and Hediger MA: The
glutamate/neutral amino acid transporter family SLC1: molecular,
physiological and pharmacological aspects. Pflugers Arch.
447:469–479. 2004. View Article : Google Scholar
|
|
21
|
Verma R, Mishra V, Sasmal D and Raghubir
R: Pharmacological evaluation of glutamate transporter 1 (GLT-1)
mediated neuroprotection following cerebral ischemia/reperfusion
injury. Eur J Pharmacol. 638:65–71. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka K, Watase K, Manabe T, et al:
Epilepsy and exacerbation of brain injury in mice lacking the
glutamate transporter GLT-1. Science. 276:1699–1702. 1997.
View Article : Google Scholar : PubMed/NCBI
|