Introduction
MicroRNAs (miRNAs) are a conserved class of small,
endogenous, non-coding single-stranded RNA molecules that have the
capacity to regulate gene expression at post-transcriptional levels
by binding to complementary sequences in the 3′ untranslated region
(3′UTR) of the target messenger RNA (mRNA), leading to mRNA
degradation or translational repression (1,2).
Evidence demonstrates that miRNAs participate in diverse biological
processes, including developmental timing, cell growth, apoptosis
and patterning of the nervous system (3). Through the detection and localization
of miRNAs in genomic regions, Calin et al (4) revealed that numerous miRNAs are
located in fragile sites and genomic regions associated with
cancer, for example miR-15a and miR-16, implying that abnormal
miRNA expression may be closely associated with cancer
pathogenesis. Subsequent studies confirmed that miRNA expression is
frequently deregulated in numerous types of cancer. miR-122, a
liver-specific miRNA, is downregulated in hepatocellular carcinoma,
resulting in the suppression of cell migration and proliferation
and the induction of cell apoptosis by targeting AKT3
(5). Upregulated miR-30b/30d
contributes to melanoma cell invasion and migration by targeting
polypeptide N-acetylgalactosaminyl-transferase 7, leading to
the progression of metastasis (6).
Therefore, miRNAs are important molecules involved in the
pathogenesis of numerous types of cancer and have potential as key
biomarkers for cancer diagnosis or prognosis (1).
Colorectal cancer (CRC) is one of the most prevalent
malignancies worldwide and remains the second leading cause of
malignancy-associated mortality, with one-third of patients
succumbing to this disease (7).
CRC often occurs due to lifestyle and increasing age, though a
minority of the population have a greater susceptibility due to
genetic mutations of tumor suppressors, oncogenes or the miRNA
network (8,9). With the development of novel
therapeutic strategies for patients with CRC, incidence has
declined in the United States of America, although incidence is
still increasing in certain countries (10). Previous studies have indicated that
numerous miRNAs are differentially expressed in human CRC tissues,
compared with those of adjacent non-cancerous tissues by microarray
assay and reverse transcription quantitative polymerase chain
reaction (RT-qPCR). These miRNAs include miR-96, miR-135b, miR-31
and miR-183, which are also upregulated in colorectal cancer
tissues (11,12). The functions of miR-135b, miR-31
and miR-183 in colorectal carcinogenesis indicated that these
miRNAs promoted CRC growth and metastasis-associated traits by
functioning as oncogenes (13–15).
Therefore, further study was required in order to elucidate the
roles and regulatory mechanisms of miR-96 in CRC.
The present study aimed to confirm whether miR-96
was overexpressed in CRC and evaluate the effects of miR-96
overexpression in CRC. miR-96 expression in 20 CRC tissues and
corresponding non-cancerous tissues was determined using RT-qPCR.
Functional studies, including MTT assay, colony formation assay and
cell cycle progression were used to analyze the effects of miR-96
overexpression. The present study additionally aimed to identify
the target genes of miR-96 in CRC using western blot analysis and
luciferase assay. The results of the present study may aid the
development of miRNA-based therapies for the treatment of CRC.
Materials and methods
Patient tissue samples
Twenty CRC tissues and corresponding non-cancerous
tissues were acquired from patients primarily diagnosed with CRC by
pathological confirmation and immunohistochemical staining at the
People’s Hospital of Weifang (Weifang, China). The samples were
obtained during surgery. Consent was obtained from all patients for
tissue sample collection and the study was approved by the Ethics
Committee of the People’s Hospital of Weifang (Weifang, China).
Following collection, samples were stored at −80°C prior to miR-96
expression analysis.
Cell culture and transfection
The human CRC cell lines SW480 and SW620 (American
Type Culture Collection, Manassas, VA, USA) were cultured in
RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco-BRL,
Invitrogen Life Technologies, Carlsbad, CA, USA). The cells were
maintained in a humidified incubator with 5% CO2 at 37°C
and digested for passage using 0.1% trypsin (Jiufeng Company,
Beijing, China). miR-96 mimics, miR-96 mimics control
(mimics-ctrl), miR-96 inhibitor and miR-96 inhibitor control were
synthesized by Ribobio Co., Ltd (Guangzhou, China). Cells were
transfected using LipofectamineTM 2000 reagent
(Invitrogen Life Technologies) according to the manufacturers’
instructions.
RNA isolation and RT-qPCR
Total RNA was extracted using TRIzol reagent
(Qiagen, Hilden, Germany) from tissues and transfected cells
according to the manufacturers’ instructions. RNA concentration and
quality was measured and 500 ng of RNA was used for reverse
transcription (RT). Moloney murine leukemia virus reverse
transcriptase (Takara Bio, Inc., Otsu, Japan) was used for the
complementary DNA (cDNA) synthesis. For the cDNA synthesis of
miR-96, an miR-96 RT primer was used and U6 small nuclear RNA was
used as an internal control. For the cDNA synthesis of target
genes, the primer Oligo(dT) was used and β-actin was used as an
internal control. Following completion of the RT reaction, the cDNA
was diluted, prepared for PCR using a SYBR Premix Ex TaqTM (Perfect
Real Time; Takara Bio, Inc.) and analyzed using an ABI 7300
Real-Time PCR System (Applied Biosystems Life Technologies, Foster
City, CA, USA). PCR was performed according to the following
conditions: 95°C for 30 sec, followed by 40 cycles at 95°C for 5
sec and 60°C for 31 sec. The stem loop RT primers were designed
according to the protocol described in a previous study (16) and the PCR primers were designed
using Primer Express® 3.0 software (Life Technologies,
Grand Island, NY, USA). The RT and PCR primers were as follows:
miR-96 RT primer, 5′-CTAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCAAAAATG-3′
and forward, 5′-ACACTCCAGCTGGGTTTGGCACTAGCACAT-3; U6 RT primer,
5′-CTCAACTGGTGTCGTGG AGTCGGCAATTCAGTTGA GAAAATATGGAAC-3′ and
forward, 5′ACACTCCAGCTGG GGTGCTCGCTTCGGCAGCACA-3′; β-actin forward,
5′-CTCCATCCTGG CCTCGCTGT-3′ and reverse, 5′-GCTGTCACCTTCACCGTT
CC-3′; tumor protein p53 inducible nuclear protein 1 (TP53INP1)
forward, 5′-CTGTGCATAACTCCTGCCCT-3′ and reverse,
5′-AACAATGAATATGCTGCCCC-3′; forkhead box protein O1 (FOXO1)
forward, 5′-GAGTGGATGGTCAA GAGCGT-3′ and reverse,
5′-TTCCTTCATTCTGCACACGA-3′; FOXO3a forward,
5′-CTACGAGTGGATGGTGCGTT-3′ and reverse,
5′-TCTTGCCAGTTCCCTCATTC-3′.
MTT assay
Cell viability was determined by MTT assay. Briefly,
transfected cells were seeded into 96-well plates and 10 μl MTT
(concentration, 5 mg/ml; Sigma-Aldrich, St Louis, MO, USA) was
added into 100 μl medium at indicated time-points. The cells were
incubated with MTT for ~4 h at 37°C, followed by removal of MTT and
addition of 150 μl DMSO. Following incubation with DMSO for 10 min
in the dark, absorbance was measured at 570 nm (A570nm)
with a microplate reader (Biorad-168-1000XC; Bio-Rad Laboratories,
Hercules, CA, USA).
Colony formation assay
Transfected cells were seeded at a density of 200
cells/well into 12-well plates for the colony formation assay. The
medium was replaced every three days until the majority of the
colonies contained >50 cells. The colonies were washed with
phosphate-buffered saline (PBS), fixed with 4% formalin and stained
with 5% crystal violet (Jinglai Company, Shanghai, China) for ~15
min. Finally, the colonies were imaged (Nikon-D610; Nikon
Corporation, Tokyo, Japan) and counted.
Cell cycle analysis
Cell cycle analysis was performed using propidium
iodide (PI) staining and flow cytometric analysis. The transfected
cells were starved for 24 h, followed by the addition of medium
containing 10% FBS for a further 24 h. The cells were subsequently
trypsinized and resuspended in cold PBS (0.1% FBS). Following
centrifugation at 228 × g for 5 min, the cells were washed twice
with cold PBS (0.1% FBS) and fixed in 70% ethanol for a minimum of
one day. Following fixation, the cells were washed with cold PBS
(0.1% FBS) and the cell pellets were resuspended in 500 μl PI
solution [50 μg/ml PI (Dingguo Biotechnology Co., Ltd., Beijing,
China) from 50X stock solution (2.5 mg/ml), 0.1 mg/ml RNase A and
0.05% Triton X-100] for ~40 min at 37°C. Finally, 3 ml cold PBS was
added and the cells were centrifuged at 513 × g for 5 min. The cell
pellets were resuspended in 500 μl PBS for flow cytometry (BD
FACSCalibur™ Cell Sorting system; BD Biosciences, Franklin Lakes,
NJ, USA).
Western blot analysis
Western blot analysis was used to evalute TP53INP1,
FOXO1 and FOXO3a protein expression in the transfected cells.
Briefly, the cells were collected and lysed with
radioimmunoprecipitation assay buffer (50 mm Tris, pH 7.4, 150 mm
NaCl, 1% nonyl phenoxypolyethoxylethanol, 0.5% sodium deoxycholate
and 0.1% SDS) for 10 min on ice. Following centrifugation at 8,000
× g for 30 min at 4°C, the supernatant was removed and the protein
concentration was measured by bicinchoninic acid assay. 50 μg
protein was analyzed using 10% SDS-PAGE. Following gel separation,
the proteins were transferred into the polyvinylidene difluoride
membrane (Dingguo Biotechnology Co., Ltd.) and blocked in 5% milk.
Rabbit polyclonal TP53INP1 antibody (ab9775; dilution, 1:1,000;
Abcam, Cambridge, UK), rabbit monoclonal FOXO1 antibody (ab52587,
1:1,000 dilution; Abcam) and rabbit polyclonal FOXO3a antibody
(ab47409; dilution, 1:1,000; Abcam) were used as primary
antibodies. The horseradish peroxidase-conjugated goat monoclonal
to rabbit immunoglobulin G was used as the secondary antibody
(Sanying Biotechnology Inc., Wuhan, China). The bound antibodies
were detected using Enhanced Chemiluminescence Plus Western
Blotting Detection system (GE Healthcare, Little Chalfont, UK).
Luciferase reporter assay
The direct interaction between miRNA and its target
mRNA was determined by luciferase reporter assay. The binding site
of miR-96 in the 3′UTR of target mRNA was cloned into the pmirGLO
Dual-Luciferase miRNA Target Expression Vector (Promega Corp.,
Madison, WI, USA) according to the manufacturer’s instructions. The
mutant binding sites were generated using Quikchange Lightning
SiteDirected Mutagenesis kit (Agilent, Technologies, Inc., Santa
Clara, CA, USA) according to the manufacturer’s instructions. Cells
were subsequently co-transfected with miRNA and the wild-type or
mutant 3′UTR for luciferase assay. Following transfection for 48 h,
the cells were harvested for protein extraction. Luciferase
intensity was examined using the Dual Luciferase Reporter Gene
Assay kit (Beyotime Institute of Biotechnology, Haimen, China)
according the manufacturer’s instructions. Renilla
luciferase intensity was used an internal control.
Statistical analysis
Values are presented as the mean ± standard
variation of three independent experiments. The differences between
groups were examined by two paired Student’s t-test (GraphPad
version 5.0; GraphPad Software, Inc., La Jolla, CA, USA) and
P<0.05 was considered to indicate a statistically significant
difference between values.
Results
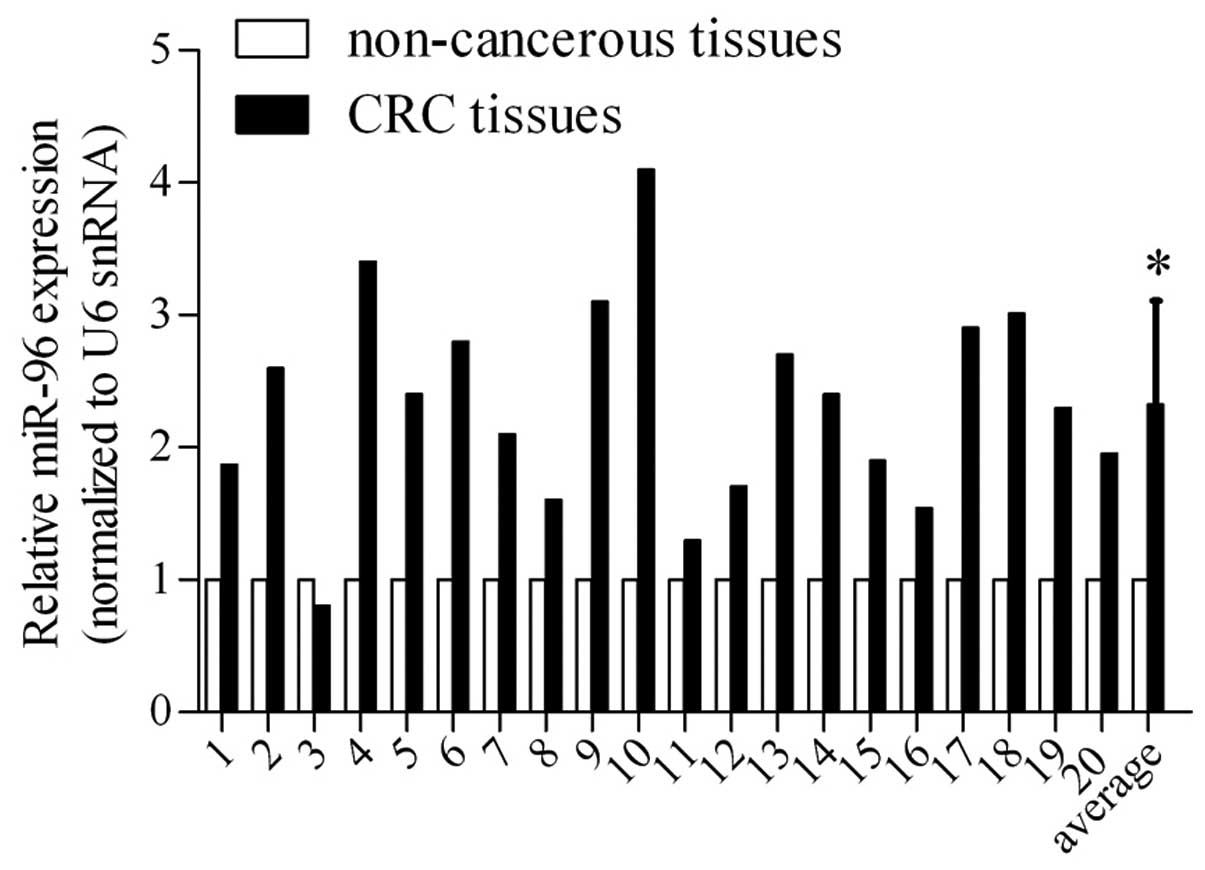
miR-96 is upregulated in CRC tissues
Based on the results of previous studies, which
indicated deregulated miRNA expression in CRC tissues and adjacent
non-cancerous tissues as indicated by microarray assay (11,12),
miR-96 expression levels were examined using RT-qPCR in 20 paired
CRC tissues and corresponding non-cancerous tissues (Fig. 1). The results demonstrated miR-96
was overexpressed in the majority of the 20 CRC tissues and
revealed that the mean expression levels were ~1.5-fold greater
than those in the corresponding non-cancerous tissues, suggesting
that the high-level expression of miR-96 may be correlated with CRC
pathogenesis.
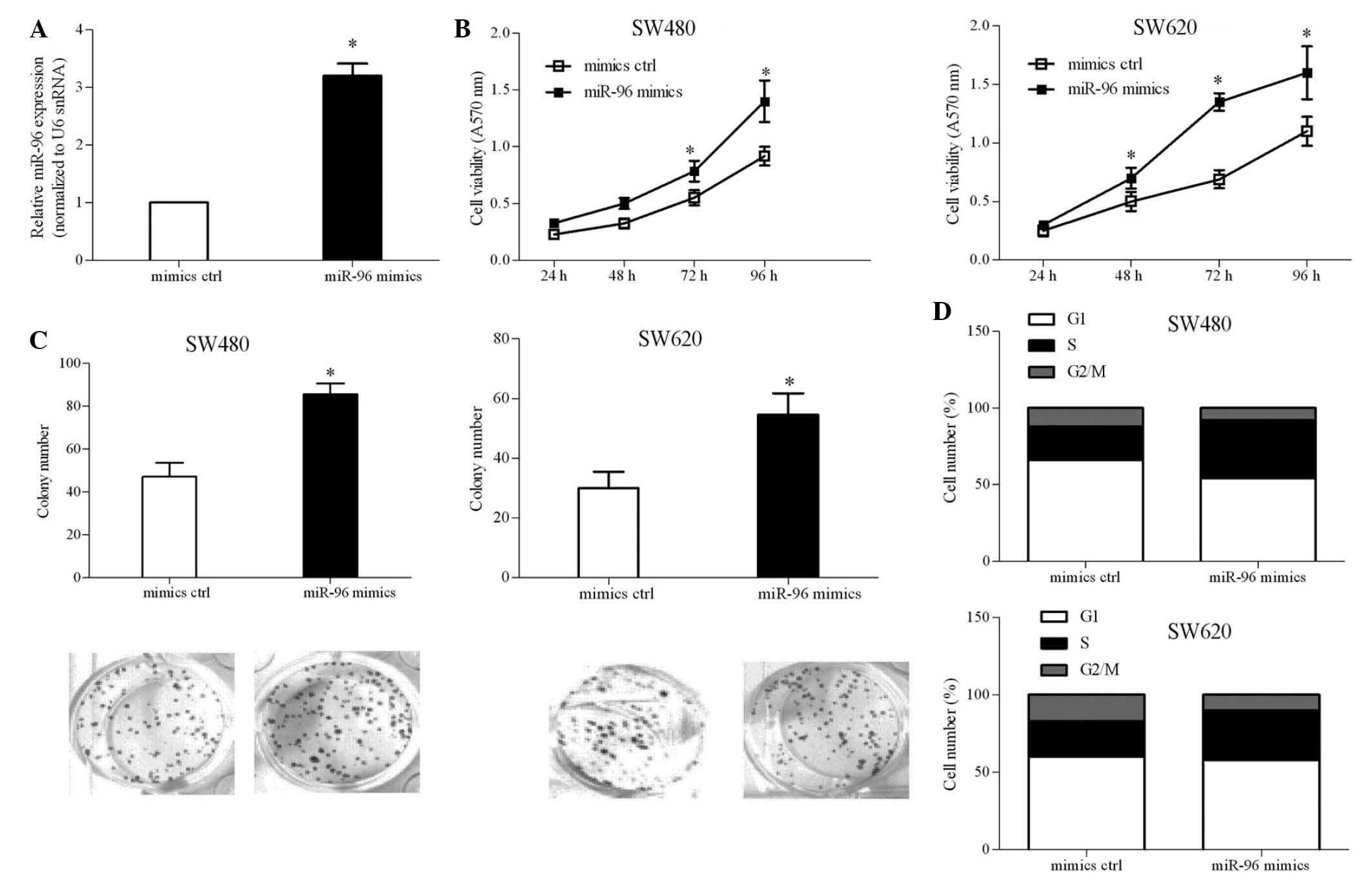
miR-96 overexpression promotes CRC cell
proliferation
To investigate the functional significance of
deregulated miR-96 expression in the CRC cellular process, ectopic
expression of miR-96 was achieved in SW480 and SW620 cells by
transient transfection with miR-96 mimics (Fig. 2A). Cell viability was investigated
at the indicated time-points by MTT assay. As indicated in Fig. 2B, it was demonstrated that ectopic
miR-96 expression increased SW480 cell viability by ~15%, compared
with that of the mimics ctrl-transfected cells. Ectopic miR-96
expression had the same effect on SW620 cell viability. A colony
formation assay was performed to determine the roles of miR-96 in
CRC-cell colony formation. It was discovered that miR-96 increased
the number of colonies by ~50 and ~80% in SW480 and SW620 cells,
respectively (Fig. 2C), compared
to that of the mimics ctrl group. In order to examine whether the
contribution of miR-96 to cell growth was associated with cell
cycle progression, the cell cycle was analyzed using PI staining.
The results, shown in Fig. 2D,
indicated that the number of miR-96-transfected cells at
G1 phase was lower, whereas the number at S phase was
higher in comparison to the numbers of cells transfected with
mimics ctrl at each phase. These results suggested that miR-96
contributed to G1/S phase transition and cell
proliferation.
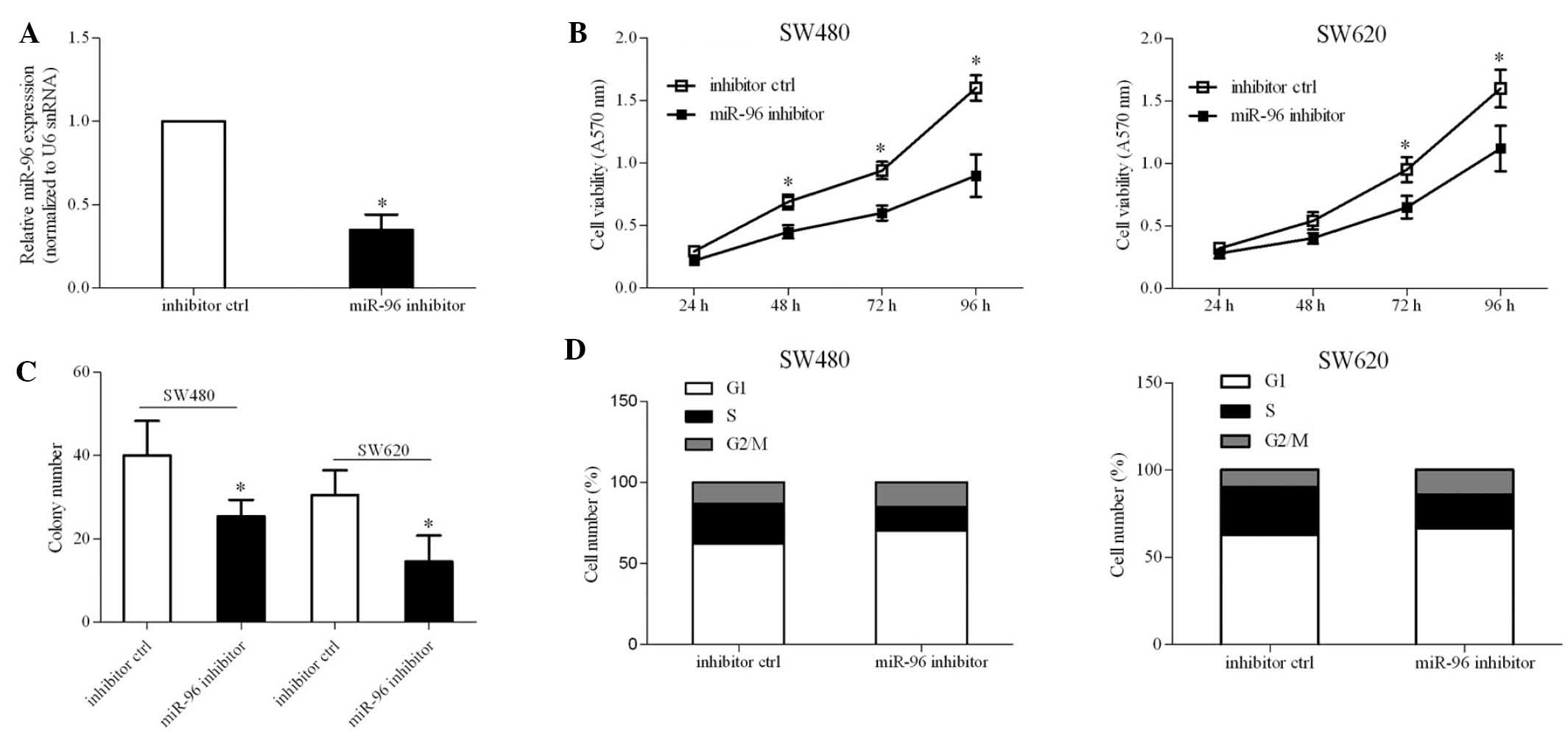
Inhibition of miR-96 results in the
suppression of CRC cell proliferation
To investigate whether miR-96 expression was
essential for CRC cell growth, endogenous miR-96 expression was
inhibited via transient transfection with an miR-96 inhibitor. The
reduction in expression of miR-96 caused by the miR-96 inhibitor
was confirmed by RT-qPCR (Fig.
3A). An MTT assay indicated that the inhibition of miR-96 led
to a ~20% reduction in cell viability (Fig. 3B). Accordingly, the number of
colonies in SW480 and SW620 cells was also reduced due to the
inhibition of miR-96 (Fig. 3C).
Cell cycle analysis revealed that miR-96 inhibition resulted in a
block of cell cycle progression at the G1 phase
transition, leading to the inhibition of cell proliferation. These
results demonstrated a significant role for miR-96 in CRC
pathogenesis.
miR-96 directly regulates TP53INP1, FOXO1
and FOXO3a expression by targeting their 3′UTR
Two algorithms, miRanda (http://www.microrna.org/microrna/home.do) and
TargetScan (http://www.targetscan.org/), were used for the
prediction of miR-96 candidate targets. Based on their potential
roles in cell growth and the miR-96 binding sites located in their
target mRNA 3′UTR sequences, TP53INP1, FOXO1 and FOXO3a were
selected as candidate targets for investigation. Previous studies
reported that FOXO1 and FOXO3a were direct targets of miR-96 in
prostate and breast cancer and were correlated with cellular
proliferation (17–19). However, the association between
miR-96 and FOXO1/FOXO3a remain to be elucidated. In the present
study, the roles of miR-96 in TP53INP1, FOXO1 and FOXO3 expression
were examined. As indicated in Fig.
4A, miR-96 inhibited the mRNA expression of TP53INP1, FOXO1 and
FOXO3a by ~30, 3 and 50%, respectively. Analogous to the effect of
miR-96 on mRNA expression levels, miR-96 also inhibited protein
expression levels of the genes evaluated (Fig. 4B).
 | Figure 4TP53INP1, FOXO1 and FOXO3a are direct
targets of miR-96. (A) Cells were transfected with miR-96 mimics or
control and harvested for RNA isolation to analyze the effect of
miR-96 on TP53INP1, FOXO1 and FOXO3a mRNA expression levels by
reverse transcription quantitative polymerase chain reaction.
β-actin was used as an internal control. (B) Transfected cells were
subjected to western blot analysis to determine protein expression
levels of TP53INP1, FOXO1 and FOXO3a. GAPDH was used as an internal
control. (C) Alignments between miR-96 and the binding sites in the
sequences at the 3′UTR. A mutation within the binding sites was
generated. The asterisks represent the mutated nucleotides. (D)
Cells were co-transfected with miR-96 and the WT or mutant 3′UTR
and subjected to luciferase reporter assay for the analysis of the
effect of miR-96 on the intensity controlled by the 3′UTR. Data
were from three independent experiments and values are expressed as
the mean ± standard deviation. *P<0.05 vs. control.
TP53INP1, tumor protein p53 inducible nuclear protein 1; FOXO1,
forkhead box protein O1; miRNA, microRNA; miR-96, microRNA-96; WT,
wild-type; mut, mutant; hsa, Homo sapiens. |
To investigate the direct regulatory roles of miR-96
in these genes, the 3′UTR-containing miR-96 binding sites (Fig. 4C) were cloned downstream of the
luciferase reporter gene. The cells were subsequently
co-transfected with miR-96 and the cloned luciferase reporter. As
indicated in Fig. 4D, it was
observed that miR-96 significantly inhibited luciferase intensity
of the 3′UTR of TP53INP1, FOXO1 and FOXO3a. To further confirm that
these binding sites mediated the inhibitory function of miR-96,
mutations within the binding sites were generated. It was
demonstrated that miR-96 did not influence the fluorescence
intensity of TP53INP1, FOXO1 and FOXO3a containing the mutant 3′UTR
(Fig. 4D). These results suggested
that TP53INP1, FOXO1 and FOXO3a were direct target genes of miR-96
and were negatively regulated by miR-96.
Discussion
miRNAs function as key regulators of gene expression
involved in diverse biological processes. A significant body of
evidence demonstrates that abnormal miRNA expression is closely
associated with the pathogenesis of numerous types of cancer
(11,12). Therefore, determination of the
differentially expressed miRNAs is crucial for elucidation of the
regulatory mechanisms of miRNAs in cancer carcinogenesis. Previous
studies have screened miRNAs using microarray and RT-qPCR for the
comparison of expression levels in CRC and those of paired
non-cancerous tissues (11,12),
and have indicated that miR-96 was significantly upregulated in CRC
tissues. miR-96 was demonstrated to promote hepatocellular cell
proliferation and colony formation (20). Studies have indicated that in
prostate cancer, miR-96 upregulation contributes to cell
proliferation and reduces cell apoptosis by targeting FOXO1
(17,21). miR-96 also enhances cell
proliferation of breast cancer by targeting FOXO1 and FOXO3a
(18,19). These results suggested that miR-96
was linked to tumor growth and carcinogenesis, functioning as an
oncogene. In agreement with the results of these previous studies,
the present study revealed that miR-96 was overexpressed in CRC
tissues, compared to expression levels in adjacent normal tissues.
Furthermore, miR-96 promoted CRC cell viability, colony formation
and cell cycle progression, while inhibition of miR-96 suppressed
cell proliferation, which indicated that miR-96 functioned as an
oncogenic miRNA.
There is disagreement regarding the potential roles
of miR-96 in diverse types of cancer. In pancreatic cancer, miR-96
was reported to be downregulated and to suppress cell
proliferation, tumor growth and metastasis-associated
characteristics by targeting the oncogene KRAS, and
therefore functioning as a tumor suppressor (22). The opposing roles of miR-96 in
cancer pathogenesis may be associated with its target genes. This
phenomenon has been reported in the regulatory effects exerted by
other miRNAs (23). For example,
miR-125b was downregulated in squamous cell carcinoma and
suppressed cell proliferation, migration and invasion by targeting
matrix metallopeptidase 13, therefore functioning as a tumor
suppressor (23). However,
miR-125b was reported to contribute to cell proliferation and
migration by the suppression of tumor suppressor TP53INP1 in type
II endometrial carcinoma cells (24). These data suggested that the
effects of miRNAs may be cell- or tissue-specific and the specific
target genes mediate their functions in disease pathogenesis.
Therefore, in the present study, the targets of miR-96 in CRC cells
were identified, which may aid in the elucidation of the regulatory
mechanisms of miR-96 in CRC.
TP53INP1 is a p53 target gene and mediates the roles
of p53 in the suppression of cell growth and induction of cell
apoptosis, functioning as a tumor suppressor (25). TP53INP1 was reported to be
downregulated in diverse types of cancer and the decreased
expression of TP53INP1 was shown to contribute to the pathogenesis
of cancer (26–28). TP53INP1 therefore has potential as
a therapeutic target for cancers. In the present study, it was
demonstrated that the TP53INP1 3′UTR contained conserved binding
sites for miR-96. RT-qPCR and western blot analysis indicated that
miR-96 inhibited TP53INP1 mRNA and protein expression. A luciferase
reporter assay was used to determine whether miR-96 directly
targeted the TP53INP1 3′UTR. The results suggested that miR-96
suppressed TP53INP1 3′UTR fluorescence intensity, whereas miR-96
had no effect on the mutant TP53INP1 3′UTR. These data suggested
that miR-96 directly suppressed TP53INP1 by binding to sequences in
the 3′UTR of TP53INP1.
In addition, two target genes of miR-96, FOXO1 and
FOXO3a, were evaluated. Previous studies have indicated that FOXO1
and FOXO3a are target genes of miR-96 in prostate and breast
cancer, respectively (17,18). FOXO1 and FOXO3a are forkhead
transcription factors of the O class, involved in cell survival,
apoptosis and the suppression of tumor growth (29,30).
The results of the present study indicated that ectopic miR-96
expression resulted in the suppression of FOXO1 and FOXO3a mRNA and
protein expression levels. The results of the luciferase reporter
assay indicated that luciferase intensity was controlled by the
FOXO1 and FOXO3a 3′UTRs and once the 3′UTR binding sites of miR-96
were mutated, miR-96 exerted no effect on the fluorescence
intensity. These data indicated that FOXO1 and FOXO3a were target
genes for miR-96 in CRC. Considering the roles of TP53INP1, FOXO1
and FOXO3a and also the regulation of their expression by miR-96,
it was hypothesized that these genes mediated the functions of
miR-96 in CRC cell growth and proliferation. Further study is
required in order to elucidate the therapeutic potential of these
genes in CRC.
In conclusion, the results of the present study
indicated that miR-96 was upregulated in CRC and promoted cell
growth and proliferation, functioning as an oncogene. In addition,
the direct target genes of miR-96 in CRC, TP53INP1, FOXO1 and
FOXO3a were identified, which may aid in the elucidation of the
regulatory mechanisms of miR-96 in CRC carcinogenesis. These
results suggested that miR-96 may act as a biomarker for CRC
diagnosis and that it has potential as an miRNA-based therapeutic
target for the treatment of CRC. Future studies should focus on
in vivo investigation of the functions of miR-96 in CRC in
order to provide more beneficial evidence for the clinical
application of miRNAs.
References
|
1
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brennecke J, Stark A, Russell RB and Cohen
SM: Principles of microRNA-target recognition. PLoS Biol.
3:e852005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calin GA, Sevignani C, Dumitru CD, et al:
Human microRNA genes are frequently located at fragile sites and
genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nassirpour R, Mehta PP and Yin MJ: miR-122
regulates tumorigenesis in hepatocellular carcinoma by targeting
AKT3. PLoS One. 8:e796552013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gaziel-Sovran A, Segura MF, Di Micco R, et
al: miR-30b/30d regulation of GalNAc transferases enhances invasion
and immunosuppression during metastasis. Cancer Cell. 20:104–118.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar
|
|
8
|
Kanthan R, Senger JL and Kanthan SC:
Molecular events in primary and metastatic colorectal carcinoma: a
review. Patholog Res Int. 2012:5974972012.PubMed/NCBI
|
|
9
|
Kaemmerer E, Klaus C, Jeon MK and Gassler
N: Molecular classification of colorectal carcinomas: The
genotype-to-phenotype relation. World J Gastroenterol.
19:8163–8167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sarver AL, French AJ, Borralho PM, et al:
Human colon cancer profiles show differential microRNA expression
depending on mismatch repair status and are characteristic of
undifferentiated proliferative states. BMC Cancer. 9:4012009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bandres E, Cubedo E, Agirre X, et al:
Identification by real-time PCR of 13 mature microRNAs
differentially expressed in colorectal cancer and non-tumoral
tissues. Mol Cancer. 5:292006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sarver AL, Li L and Subramanian S:
MicroRNA miR-183 functions as an oncogene by targeting the
transcription factor EGR1 and promoting tumor cell migration.
Cancer Res. 70:9570–9580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Khatri R and Subramanian S: MicroRNA-135b
and its circuitry networks as potential therapeutic targets in
colon cancer. Front Oncol. 3:2682013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cekaite L, Rantala JK, Bruun J, et al:
MiR-9, -31, and -182 deregulation promote proliferation and tumor
cell survival in colon cancer. Neoplasia. 14:868–879.
2012.PubMed/NCBI
|
|
16
|
Chen C, Ridzon DA, Broomer AJ, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haflidadottir BS, Larne O, Martin M, et
al: Upregulation of miR-96 enhances cellular proliferation of
prostate cancer cells through FOXO1. PLoS One. 8:e724002013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin H, Dai T, Xiong H, et al: Unregulated
miR-96 induces cell proliferation in human breast cancer by
downregulating transcriptional factor FOXO3a. PLoS One.
5:e157972010. View Article : Google Scholar
|
|
19
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast
cancer cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu D, He X, Chang Y, et al: Inhibition of
miR-96 expression reduces cell proliferation and clonogenicity of
HepG2 hepatoma cells. Oncol Rep. 29:653–661. 2013.
|
|
21
|
Fendler A, Jung M, Stephan C, et al: The
antiapoptotic function of miR-96 in prostate cancer by inhibition
of FOXO1. PLoS One. 8:e808072013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu S, Lu Z, Liu C, et al: miRNA-96
suppresses KRAS and functions as a tumor suppressor gene in
pancreatic cancer. Cancer Res. 70:6015–6025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu N, Zhang L, Meisgen F, et al:
MicroRNA-125b down-regulates matrix metallopeptidase 13 and
inhibits cutaneous squamous cell carcinoma cell proliferation,
migration, and invasion. J Biol Chem. 287:29899–29908. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang F, Liu T, He Y, et al: MiR-125b
promotes proliferation and migration of type II endometrial
carcinoma cells through targeting TP53INP1 tumor suppressor in
vitro and in vivo. BMC Cancer. 11:4252011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang CM, Zhao J and Deng HY: MiR-155
promotes proliferation of human breast cancer MCF-7 cells through
targeting tumor protein 53-induced nuclear protein 1. J Biomed Sci.
20:792013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang PH, Motoo Y, Garcia S, et al:
Down-expression of tumor protein p53-induced nuclear protein 1 in
human gastric cancer. World J Gastroenterol. 12:691–696.
2006.PubMed/NCBI
|
|
27
|
Shahbazi J, Lock R and Liu T: Tumor
protein 53-induced nuclear protein 1 enhances p53 function and
represses tumorigenesis. Front Genet. 4:802013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ito Y, Motoo Y, Yoshida H, et al:
Decreased expression of tumor protein p53-induced nuclear protein 1
(TP53INP1) in breast carcinoma. Anticancer Res. 26:4391–4395.
2006.
|
|
29
|
Maiese K, Chong ZZ, Shang YC and Hou J:
Clever cancer strategies with FoxO transcription factors. Cell
Cycle. 7:3829–3839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo JP, Tian W, Shu S, et al: IKBKE
phosphorylation and inhibition of FOXO3a: a mechanism of IKBKE
oncogenic function. PLoS One. 8:e636362013. View Article : Google Scholar : PubMed/NCBI
|