Introduction
Gastric cancer is the fourth most common type of
cancer and the second leading cause of cancer-associated mortality
worldwide (1), with approximately
one million new cases diagnosed each year. The phosphoinositide-3
kinases (PI3Ks) are a family of enzymes that have been categorized
into three classes, class I, II and III. Class III PI3K is
constitutively active and is able to generate phosphatidylinositol
3-phosphate from phosphatidylinositol (2–4).
Various studies have shown that the class III phosphatidylinositol
3-kinase signaling pathways are the central conduit in the
regulation of autophagy (5).
Previous studies have indicated that autophagy is
involved in tumor cell resistance to chemotherapy and that
inhibition of autophagy enhances the cytotoxicity of certain
chemotherapeutic agents (6,7).
Autophagy is an important mechanism in various physiopathological
processes, including tumorigenesis, development, cell death and
survival (8,9). Autophagy has also been demonstrated
to have a complex association with apoptosis, with an important
role in promoting cell survival against apoptosis (10). The induction of cell death and the
inhibition of cell survival are the main principles of cancer
therapy. Resistance to chemotherapeutic agents is a major problem
in oncology and limits the efficacy of anticancer drugs.
The identification of Class III PI3K revealed a
novel role for autophagy in inducing cell death, and Class III PI3K
is considered to be a crucial modulator of apoptosis and autophagy.
In the present study, the effects of PI3K(III)-RNA interference
(i)-green fluorescent protein (GFP) adenovirus (AD) on the growth
and apoptosis of gastric cancer cells in vitro were
analyzed. The effects of the PI3K(III)-RNAi-GFP AD on the
activation of apoptosis and autophagy, and the contribution of
autophagy to chemosensitivity effects of 5-fluorouracil (5-FU) in
the SGC7901 gastric cancer cell line were investigated.
Materials and methods
Reagents
SGC7901 gastric cancer cells were obtained from the
Shanghai Institute of Cell Biology, Chinese Academy of Sciences
(Shanghai, China). RPMI-1640 medium was provided by Gibco-BRL
(Rockville, MD, USA). Fetal calf serum (FCS) was purchased from
Hangzhou Sijiqing Biological Engineering Material Co., Ltd.
(Hangzhou, China). L-glutamine and methyl thiazolyl tetrazolium
(MTT) were provided by Sigma-Aldrich (St. Louis, MO, USA).
Adenoviral vectors and infections
An RNAi sequence against Class III PI3K was designed
and vectors that expressed Class III PI3K small interfering (si)RNA
were constructed. A Class III PI3K-specific target sequence was
selected with the Invitrogen online siRNA tool (http://www.invitrogen.com/rnai) using the Class
III PI3K reference sequence (GenBank accession no. NM_002647). The
target sequence was as follows: Class III PI3K (base 2521–2641),
5′-TCCGCTTAGACCTGTCGGATGAAGAGGCT-3′. Subsequently, the siRNA was
chemically synthesized and a lentiviral vector was constructed. The
insertion of the specific siRNA was further verified by sequencing.
The siRNA were sequenced on the SOLiD 5500xl system (35 bp read
length; Shanghai Genesil Co., Ltd., Shanghai, China). The
recombinant AD vector that expresses siRNA against Class III PI3K
was synthesized by Shanghai Genesil Co., Ltd. The
replication-defective adenoviral vectors expressing GFP
(PI3K(III)-RNAi-GFP-AD and control negative control
(NC)-RNAi-GFP-AD) were stored at −80°C. Infections were performed
at 70–75% confluence in Dulbecco’s modified Eagle’s medium
supplemented with 2% FCS. The cells were subsequently incubated at
37°C for a minimum of 4 h, followed by the addition of fresh
medium. The cells were subjected to functional analysis at fixed
time points following infection as described for the individual
experimental conditions.
Drug preparation
5-FU (Sigma-Aldrich) was diluted in saline to
produce a stock solution that was stored according to the
manufacturers’ instructions. The 5-FU concentration selected for
use in the experiments was 2 mg/l, in accordance with the
manufacturer’s instructions.
Determination of optimal multiplicity of
infection (MOI)
A total of 1×104 SGC7901 cells/well were
seeded in 96-well plates to produce 60–70% cultured adherent cells.
Subsequently, 10, 20, 30, 50 and 100 MOI of the NC-RNAi-GFP-AD was
added to 100 μl diluted infected cells, 8 h after the addition of
RPMI 1640 culture medium with 10% fetal bovine serum. The cells
were cultured for 48 h then counted using a fluorescence microscope
(Leica DMI4000B; Leica Microsystems Wetzlar GmbH, Wetzlar, Germany)
to calculate the number of cells that expressed GFP.
Cell culture and MTT assay
The SGC7901 cells were maintained in RPMI 1640
medium containing 10% heat-inactivated fetal bovine serum and 0.03%
L-glutamine, and were incubated in a 5% CO2 atmosphere
at 37°C. Cell viability was evaluated using an MTT assay. To
analyze the response to PI3K(III)-RNAi-GFP-AD, the cells were
plated into 96-well microplates (7×104 cells/well) and
the PI3K(III)-RNAi-GFP-AD was added to the culture medium. MTT
solution was then added to the culture medium (500 μg/ml final
concentration) for 4 h prior to the end of treatment and the
reaction was terminated by the addition of 10% acidic sodium
dodecyl sulfate (100 μl). The optical density of each well was
determined using a microculture plate reader (Bio-Rad Laboratories,
Richmond, CA, USA) at 570 nm. Each absorbance value was normalized
to the survival percentage value. Each assay was performed in
triplicate.
Visualization of monodansylcadaverin
(MDC)-labeled autophagic vacuoles
The SGC7901 cells were plated on 24-chamber culture
slides, cultured for 24 h and then incubated with 5-FU, adenovirus
PI3K(III)-RNAi-GFP and control adenovirus NC-RNAi-GFP treatment in
RPMI 1640 with 10% FCS for 12 or 24 h. The autophagic vacuoles were
labeled with MDC (Sigma-Aldrich) as described previously (11), by incubating the cells with 0.001
mmol/l MDC in RPMI 1640 at 37°C for 10 min. Following incubation,
the cells were washed twice with phosphate-buffered saline (PBS)
and immediately analyzed under a fluorescence microscope (Nikon
Eclipse TE 300; Nikon Corporation; Tokyo, Japan). All experiments
were repeated in triplicate.
Detection of the microtubule-associated
proteins 1A/1B light chain 3A (MAP1LC3) protein by
immunofluorescence
MAP1LC3 immunodetection was performed on SGC7901
cells treated with PI3K(III)-RNAi-GFP-AD or 5-FU, and mounted onto
silanized slides. The cells were cultured on sterilized glass
coverslips, fixed with 4% paraformaldehyde and blocked with 0.1%
bovine serum albumin in PBS. The slides were then incubated with a
rabbit monoclonal anti-human antibody against LC3 (dilution, 1:150;
cat. no. 12741; Cell Signaling Technology, Inc., Beverly, MA, USA).
Following washing, the slides were incubated with a secondary ghost
monoclonal anti-rabbit cy3 immunoglobulin G (IgG)-conjugated
antibody (1:500; cat. no. SAB3700843; Sigma-Aldrich). Cell nuclei
were counter-stained with 1 μg/ml Hoechst 33258 (Keygen
Biotechnology Co., Ltd., Nanjing, China). and the slides were
treated with fluorescent mounting medium (Dako, Carpinteria, CA,
USA). Images were captured using a laser confocal microscope
(Leica, Mannheim, Germany).
Mitochondrial membrane potential
(ΔΨ)
Alterations in the mitochondrial membrane potential
were the earliest indication of cell death but not the distinction
between apoptosis and oncosis. The mitochondrial membrane potential
of the SC7901 cells was measured using a KeyGEN Mitochondrial
Membrane Sensor kit (Nanjing KeyGen Biotech. Co. Ltd., Nanjing,
China). The cells were stained and fluorescence intensity was
detected with flow cytometry (FCM; Partec, Münster, Germany).. The
mitochondrial ΔΨ was calculated from the fluorescence intensities
using CellQuest™ software (Becton Dickinson, Bedford, MA, USA)
(12).
Transmission electron microscopy
(TEM)
All reagents for these experiments were obtained
from Keygen Biotechnology Co., Ltd. For TEM, the SGC7901 cells were
fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH
7.0) for 2 h, post-fixed in 2% osmium tetroxide for 2 h and then
washed with 0.1 M cacodylate buffer. The cells were coated in 5%
Noble Agar and washed with distilled water five times, with further
fixing in 2% uranyl acetate for 2 h, followed by dehydration in 50%
(15 min), 70% (16 h), 85% (15 min), 95% (15 min) and two
incubations in 100% ethanol, 15 min each. The cells were
subsequently cleared by two incubations in propylene oxide, each 15
min, and infiltrated with epon resin:propylene oxide (1:1) for 3 h,
epon resin:propylene oxide (3:1) for 16 h and two incubations with
pure epon resin for a total of 6 h. Thin sections were mounted onto
grids and examined under an electron microscope (Philips CM120;
Koninklijke Philips N.V., Amsterdam, The Netherlands) (13).
Statistical analysis
The results are presented as the mean ± standard
deviation. Statistical analysis was performed using the Dennett’s
test and analysis of variance. P<0.05 was considered to indicate
a statistically significant difference.
Results
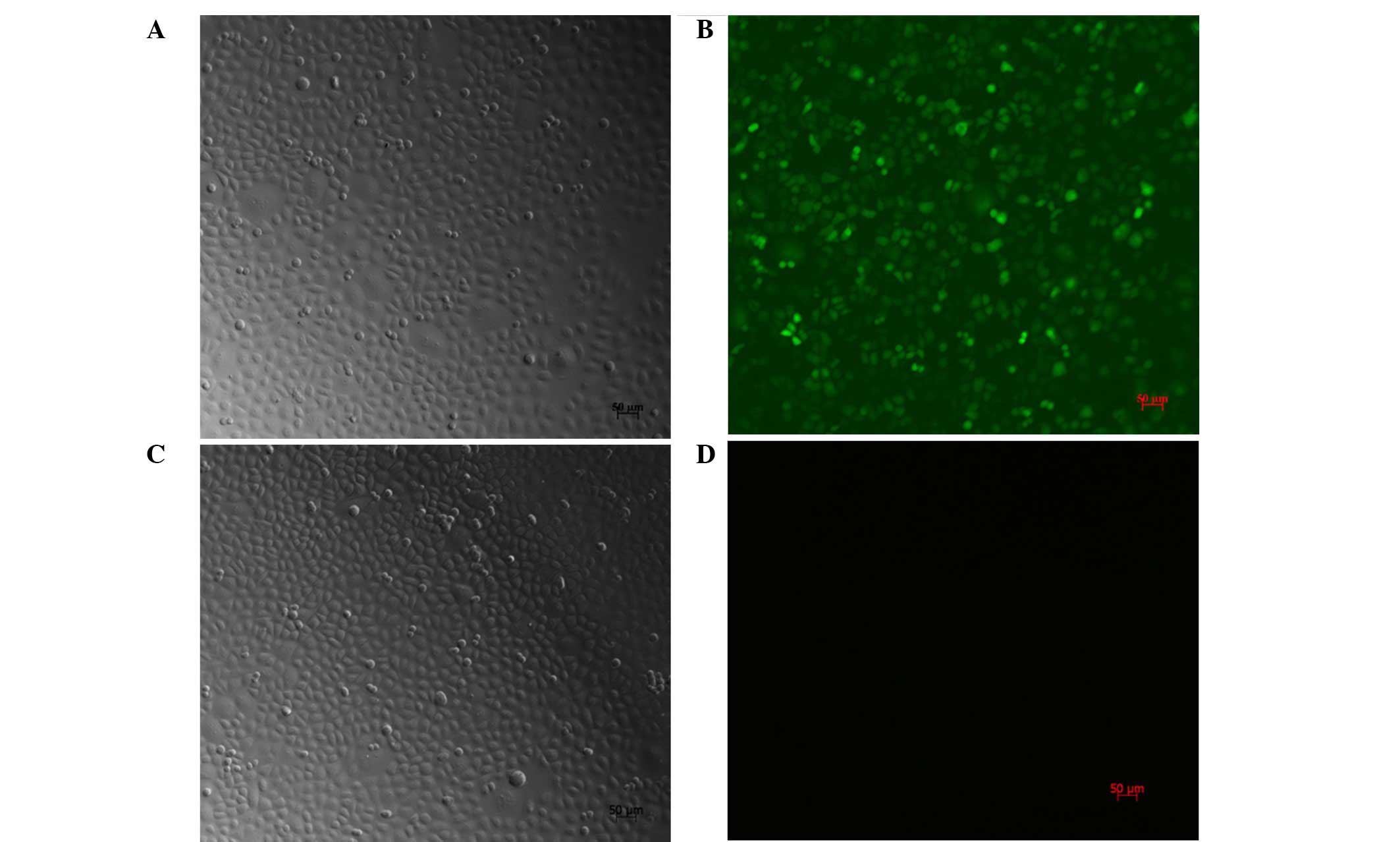
Transfection efficiency and cell
morphology
Following treatment of the SGC7901 cells with
PI3K(III)-RNAi-GFP-AD (30 MOI) for 24 h, the cell body was observed
to be swollen and rounded. The cells showed further deformation
after 48 h. Fragments by recombinant AD containing GFP, after 72 h
transfection, the SGC7901 cells were counted using a fluorescence
microscope hair green fluorescence of tumor cells (Fig. 1). At 30 MOI, the transfection
efficiency was revealed to be 95±2.4%.
PI3K(III)-RNAi-GFP-AD inhibits cell
viability and enhances 5-FU-mediated tumor cell growth
inhibition
The PI3K(III)-RNAi-GFP-AD reduced SGC7901 cell
viability in a time-dependent manner. The MTT assays revealed that
after 24 h of PI3K(III)-RNAi-GFP treatment, the rate of inhibition
reached 26.45±11.23% at 30 MOI. The rate of inhibition rose when
the incubation time was prolonged, reaching 40.58±6.54% at 48 h and
53.12%±2.0% at 72 h after treatment (Fig. 2). In order to assess the clinical
value of the PI3K(III)-RNAi-GFP-AD in tumor treatment and to
analyze the synergistic inhibitory effect of the
PI3K(III)-RNAi-GFP-AD on growth in combination with a chemotherapy
drug, 2 mg/l of the 5-FU chemotherapy drug was administered to the
cells along with the PI3K(III)-RNAi-GFP. The PI3K(III)-RNAi-GFP-AD
was demonstrated to exert a greater effect when used in combination
with 5-FU than when used alone (Fig.
2). Following the treatment of SGC7901 gastric cancer cells
with 5-FU + PI3K(III)-RNAi-AD, the absorbance values at 24, 48 and
72 h were 0.17±1.64, 0.13±4.64 and 0.11±3.56%, respectively, with
inhibition ratios at 45.89±6.67, 72.57±9.48 and 87.51±4.65%,
respectively. In the combined treatment group, as compared with
other groups, the inhibition rate was significantly higher
(P<0.05). Thus, PI3K(III)-RNAi-GFP-AD treatment inhibited the
proliferation of SGC7901 gastric cancer cells and enhanced the
chemosensitivity of the cells to 5-FU.
PI3K(III)-RNAi-GFP-AD transfection and
5-FU treatment reduce the number of autophagic vacuoles
The autofluorescent substance MDC has been recently
shown to be a marker for late autophagic vacuoles (11). To investigate the
autophagy-inhibiting effect of PI3K(III)-RNAi-GFP-AD transfection
and 5-FU treatment of SGC7901 cells, the cells were stained with
MDC. When the cells were viewed under a fluorescence microscope,
MDC-labeled autophagic vacuoles appeared as distinct dot-like
structures distributed in the cytoplasm or in the perinuclear area.
The number of MDC-labeled vesicles in the SGC7901 cells was
observed to be significantly fewer following treatment with
PI3K(III)-RNAi-GFP-AD (30 MOI) or 5-FU for between 24 and 72 h, as
compared with the number of vesicles in the control group cells
(Fig. 3). A reduction in the
number of MDC-labeled vesicles following treatment with
PI3K(III)-RNAi-GFP-AD and 5-FU was revealed, as compared with X
used alone (Fig. 3).
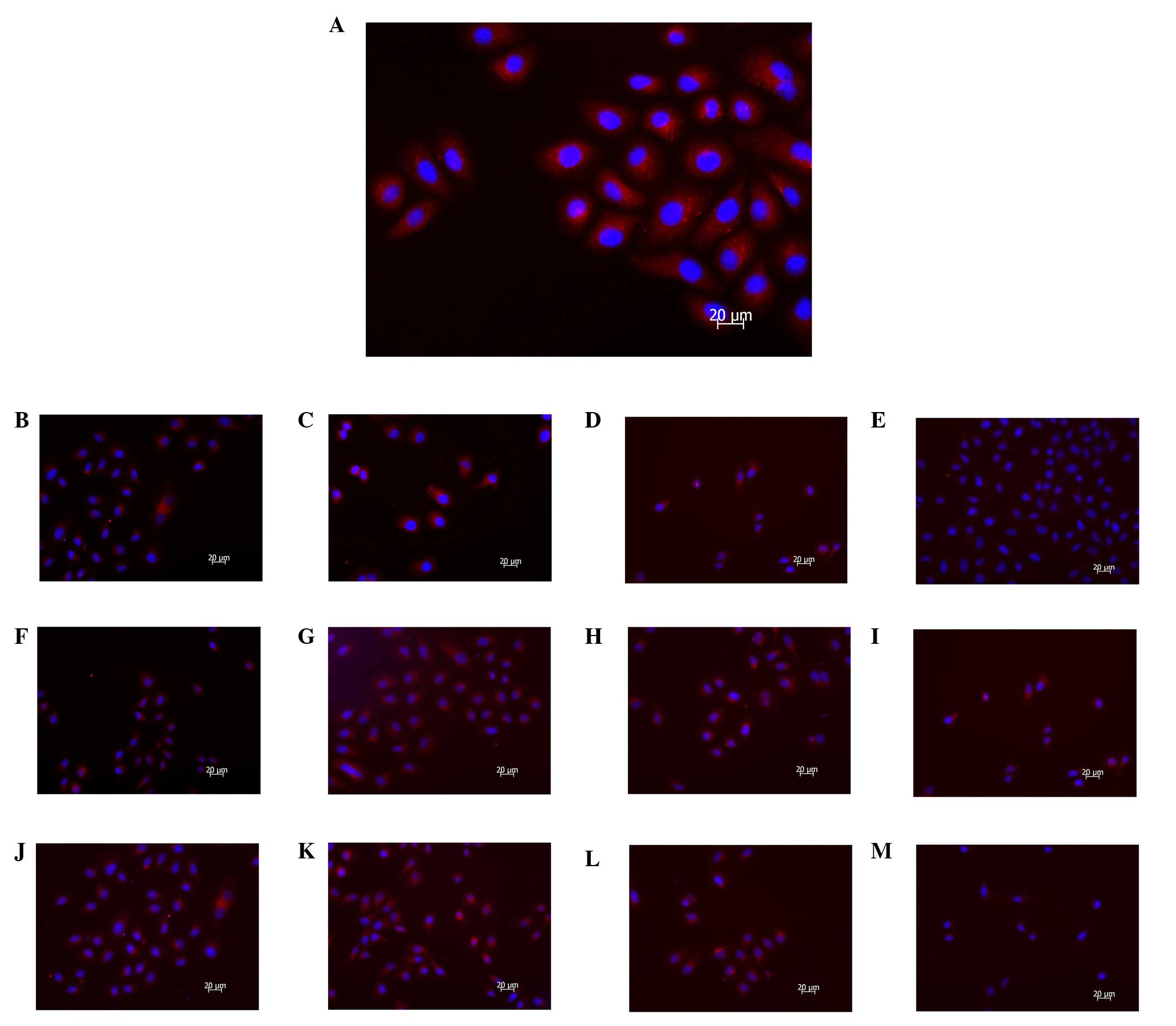
 | Figure 3MDC staining reveals that autophagy is
inhibited following PI3K(III)-RNAi-GFP adenovirus (30 MOI) or 5-FU
treatment. SGC7901 gastric cancer cells were incubated with
PI3K(III)-RNAi-GFP adenovirus (30 MOI), NC-RNAi-GFP adenovirus or
5-FU for the indicated time periods, and were then stained with MDC
(100 μmol/l). Fluorescent particles revealed late autophagic
vacuoles. (A) 24 h after control treatment. (B) 24 h after
NC-RNAi-GFP adenovirus treatment. (C) 24 h after 5-FU treatment.
(D) 24 h after PI3K(III)-RNAi adenovirus treatment. (E) 24 h after
5-FU and PI3K(III)-RNAi adenovirus treatment. (F) 48 h after
NC-RNAi-GFP adenovirus treatment. (G) 48 h after 5-FU treatment.
(H) 48 h after PI3K(III)-RNAi adenovirus treatment. (I) 48 h after
5-FU and PI3K(III)-RNAi adenovirus treatment. (J) 72 h after
NC-RNAi-GFP adenovirus treatment. (K) 72 h after 5-FU treatment.
(L) 72 h after PI3K(III)-RNAi adenovirus treatment. (M) 72 h after
5-FU and PI3K(III)-RNAi adenovirus treatment. Magnification, ×400;
n=3. MDC, monodansylcadaverin; PI3K, phosphoinositide 3-kinase;
RNAi, RNA interference; GFP, green fluorescence protein; 5-FU,
5-fluorouracil; MOI, multiplicity of infection; NC, negative
control. |
PI3K(III)-RNAi-GFP-AD transfection and
5-FU treatment reduce punctate LC3
To analyze whether autophagy was inhibited following
exposure to PI3K(III)-RNAi-GFP-AD transfection or 5-FU in SGC7901
cells, evaluation of cyanine 3 (CY3)-LC3 modulation using
fluorescent microscopic methods was employed. LC3 is a mammalian
homologue of autophagy-related protein 8 (Atg8), a protein required
for autophagy that is recruited to the autophagosomal membrane,
which is routinely examined to monitor autophagy. CY3-LC3, LC3
fused to red fluorescent protein, measured as an elevation in
punctate CY3-LC3, was detected with fluorescence microscopy. The
results revealed that the PI3K(III)-RNAi-GFP-AD and 5-FU treatments
reduced the punctate distribution of LC3 immunoreactivity, which
indicated downregulation of autophagosome formation by
PI3K(III)-RNAi-GFP-AD between 24 and 72 h (Fig. 4). The immune fluorescence
expression levels of LC3, which is a specific autophagy protein,
were significantly downregulated, as compared with other groups, in
the combined treatment group, as determined by fluorescence
microscopy. A greater inhibition of LC3 punctate distribution was
observed following treatment with PI3K(III)-RNAi-GFP-AD and 5-FU
than when X was used alone (Fig.
4).
PI3K(III)-RNAi-GFP-AD transfection and
5-FU treatment induce mitochondrial dysfunction
The mitochondrial membrane potential of the SGC7901
cells was detected using JC-1. Mitochondrial membrane potential
(ΔΨ) collapse was observed as early as 24 h after
PI3K(III)-RNAi-GFP-AD (30 MOI) treatment reaching a maximum at 72 h
(Fig. 5). The percentages of cells
with green fluorescence in the combined treatment group were
74.4±3.86 (24 h), 82.3±1.84 (48 h) and 92.5±1.1% (72 h), which were
larger than those in the respective other groups. The increase in
the percentage of cells with green fluorescence indicated that the
mitochondrial membrane potential was reduced to a greater extent.
Mitochondrial membrane potential collapse indicates cell apoptosis
or necrosis. PI3K(III)-RNAi-GFP-AD (30 MOI) induced mitochondrial
dysfunction, activated cell apoptosis and enhanced the effects of
5-FU-mediated increased green fluorescence emission in the SGC7901
cells.
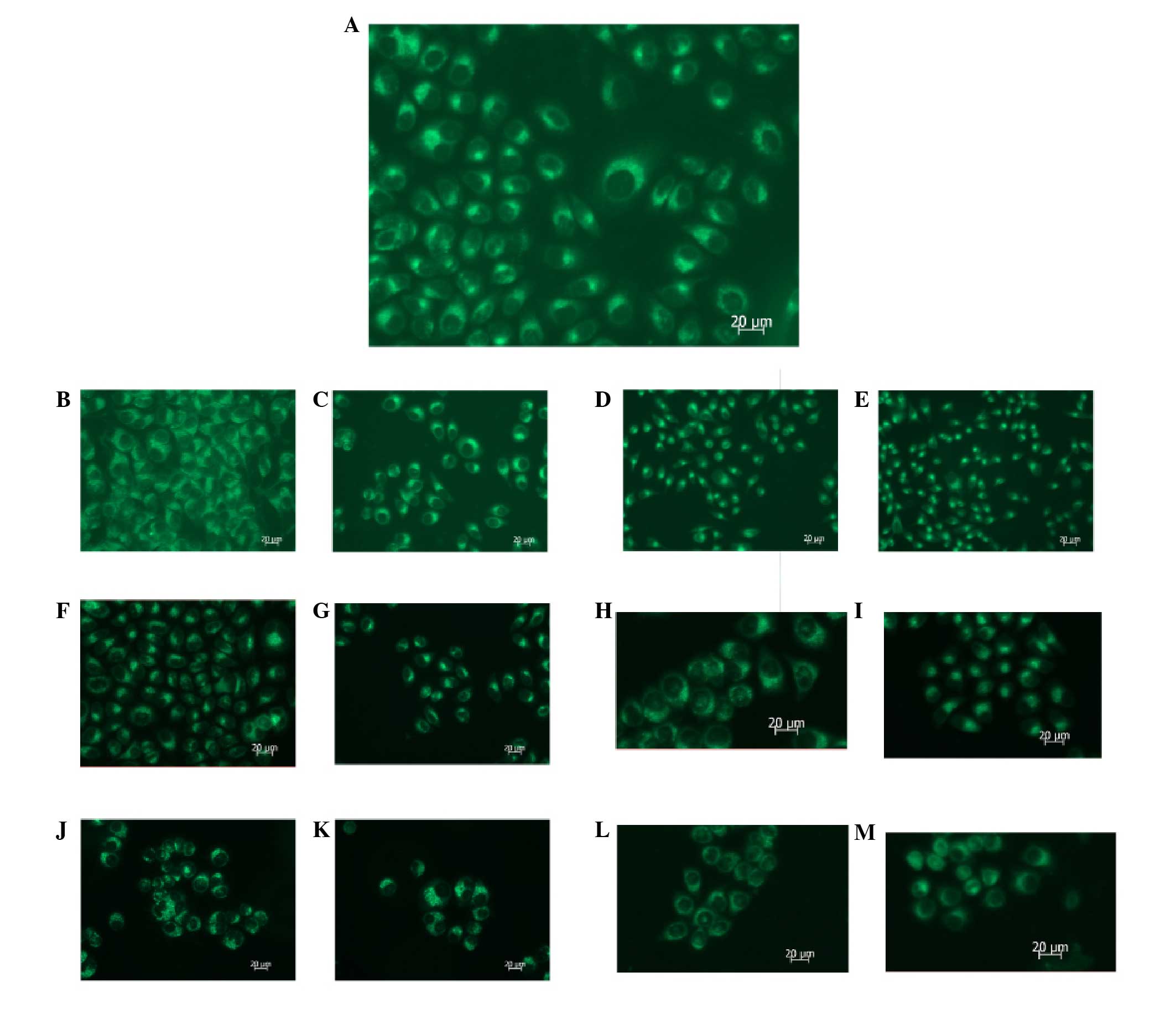
 | Figure 5Flow cytometric analysis of
mitochondrial membrane potential, indicated by green fluorescence
ratio, following treatment of SGC7901 gastric cancer cells with
PI3K(III)-RNAi-GFP-AD and/or 5-FU. The cells were incubated with
PI3K(III)-RNAi-GFP-AD (30 multiplicity of infection),
NC-RNAi-GFP-AD or 5-FU for the indicated time periods, and were
then stained with JC-1 (5 μmol/l). (A) 24 h after control
treatment. (B) 24 h after NC-RNAi-GFP-AD treatment. (C) 24 h after
5-FU treatment. (D) 24 h after PI3K(III)-RNAi-AD treatment. (E) 24
h after 5-FU and PI3K(III)-RNAi-AD treatment. (F) 48 h after
NC-RNAi-GFP-AD treatment. (G) 48 h after 5-FU treatment. (H) 48 h
after PI3K(III)-RNAi-AD treatment. (I) 48 h after 5-FU and
PI3K(III)-RNAi-AD treatment. (J) 72 h after NC-RNAi-GFP-AD
treatment. (K) 72 h after 5-FU treatment. (L) 72 h after
PI3K(III)-RNAi-AD treatment. (M) 72 h after 5-FU and
PI3K(III)-RNAi-AD treatment. N=3. PI3K, phosphoinositide 3-kinase;
RNAi, RNA interference; GFP, green fluorescent protein; 5-FU,
5-fluorouracil; NC, negative control; AD, adenovirus. |
PI3K(III)-RNAi-GFP-AD and 5-FU treatment
inhibits autophagy and impairs mitochondria
TEM was used to identify any ultrastructural changes
in the SGC7901 cells following PI3K(III)-RNAi-GFP-AD (30 MOI)
treatment. Control cells exhibited a round shape and contained
normal-looking organelles, nucleus and chromatin (Fig. 6A). However, subsequent to 5-FU
treatment, inhibition of autophagy and apoptotic body induction
were observed (Fig. 6C, G and K).
PI3K(III)-RNAi-GFP-AD (30 MOI)-treated cells also exhibited
inhibited autophagy and induced cell apoptosis, as shown in
Fig. 6D, H and J. Following 5-FU
and PI3K(III)-RNAi-GFP-AD (30 MOI) combined treatment, the effects
of 5-FU-induced cell death were elevated (Fig. 6E, I and M). The loss of organelles
and cytoplasm vacuolization was also observed when the incubation
time was prolonged. The degree of autophagy in the combined
treatment group was significantly reduced, as compared with the
other treatment groups.
Discussion
In the present study, the small interfering RNA
(siRNA) of class III PI3K was revealed to reduce viability, inhibit
autophagy and induce apoptosis in SGC7901 gastric cancer cells,
thus demonstrating the cytotoxic effects of the
PI3K(III)-RNAi-GFP-AD. These findings indicate that using siRNA
targeting molecules in the class III PI3K signaling pathway is a
potential strategy for treating gastric cancer. In addition,
PI3K(III)-RNAi-GFP-AD was also observed to enhance the effects of
5-FU in inducing cell death.
Resistance to anticancer drug-induced cell death is
a predominant cause of cancer treatment failure. The majority of
the chemotherapeutic agents kill cancer cells via the mitochondrial
pathway. The mitochondrial pathway of apoptosis exerts a critical
role. Mitochondrial permeability transition is an important event
in the initiation of apoptosis (14). The findings from the present study
revealed that the mitochondrial ΔΨ collapsed following
PI3K(III)-RNAi-GFP-AD treatment. Mitochondrial dysfunction is
considered to be one of the most common and consistent phenotypes
of cancer cells.
The majority of studies have indicated that
autophagic inhibition sensitizes tumor cells to a wide spectrum of
cancer therapies, while others have observed that treatment-induced
tumor cell death requires the autophagic pathway (15). Autophagic inhibition may snergize
with other chemotherapeutic mechanisms to more effectively
eliminate cancer cells. Recent studies supported this hypothesis,
demonstrating promising effects in response to diverse
chemotherapeutic agents, highlighting the possibility of
autophagy-targeting adjuvant therapy in the treatment of cancer.
The effects of autophagic inhibition treatment administered in
combination with current anticancer therapies has been previously
examined in various tumor models (16–18).
In the present study, PI3K(III)-RNAi-GFP-AD exerted a greater
effect when used in combination with 5-FU than when used alone.
Thus, PI3K(III)-RNAi-GFP-AD inhibited the proliferation of SGC7901
gastric cancer cells, suppressed cell autophagy, induced cell
apoptosis and enhanced the chemosensitivity of 5-FU.
In conclusion, autophagic inhibition and apoptotic
activation may markedly contribute to PI3K(III)-RNAi-GFP-AD-induced
SGC7901 cell death. In addition, autophagic inhibition may
sensitize the cell chemosensitivity to 5-FU. Further investigation
of upstream signal regulation of autophagy and apoptosis may
provide novel insights with regard to the underlying mechanisms
accommodating or contributing to autophagy and apoptosis, thereby
unveiling novel strategies for tumor therapy.
Acknowledgements
This study was supported by the Natural Science
Foundation of China (grant no. 81172348) and the Suzhou Science and
Technology Development Foundation (grant nos. 2010SYS201031 and
2011SYSD2011092).
References
|
1
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.PubMed/NCBI
|
|
2
|
Wu H, Yan Y and Backer JM: Regulation of
class IA PI3Ks. Biochem Soc Trans. 35:242–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yan Y and Backer JM: Regulation of class
III (Vps34) PI3Ks. Biochem Soc Trans. 35:239–241. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abedin MJ, Wang D, McDonnell MA, Lehmann U
and Kelekar A: Autophagy delays apoptotic death in breast cancer
cells following DNA damage. Cell Death Differ. 14:500–510. 2007.
View Article : Google Scholar
|
|
7
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Biederbick A, Kern HF and Elsässer HP:
Monodansylcadaverine (MDC) is a specific in vivo marker for
autophagic vacuoles. Eur J Cell Biol. 66:3–14. 1995.PubMed/NCBI
|
|
12
|
Hu Y, Xia XY, Pan LJ, et al: Evaluation of
sperm mitochondrial membrane potential in varicocele patients using
JC-1 fluorescent staining. Zhonghua Nan Ke Xue. 15:792–795.
2009.(In Chinese). PubMed/NCBI
|
|
13
|
Kapp OH, Mainwaring MG, Vinogradov SN and
Crewe AV: Scanning transmission electron microscopic examination of
the hexagonal bilayer structures formed by the reassociation of
three of the four subunits of the extracellular hemoglobin of
Lumbricus terrestris. Proc Natl Acad Sci USA. 84:7532–7536. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Biswas G, Guha M and Avadhani NG:
Mitochondria-to-nucleus stress signaling in mammalian cells: Nature
of nuclear gene targets, transcription regulation, and induced
resistance to apoptosis. Gene. 354:132–139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Degtyarev M, De Mazière A, Orr C, et al:
Akt inhibition promotes autophagy and sensitizes PTEN-null tumors
to lysosomotropic agents. J Cell Biol. 183:101–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lomonaco SL, Finniss S, Xiang C, et al:
The induction of autophagy by gamma-radiation contributes to the
radioresistance of glioma stem cells. Int J Cancer. 125:717–722.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shingu T, Fujiwara K, Bögler O, et al:
Stage-specific effect of inhibition of autophagy on
chemotherapy-induced cytotoxicity. Autophagy. 5:537–539. 2009.
View Article : Google Scholar : PubMed/NCBI
|