Introduction
Glioblastoma multiforme (GBM) is one of the most
common and aggressive forms of brain tumor, accounting for 50–60%
of all brain cancers in humans, and is associated with a low median
survival rate (1). GBM is
generally characterized by high lethality, invasiveness, excessive
growth, and a poor prognosis (2,3). The
current gold-standard treatment strategy for brain tumors is
comprised of radiation therapy, surgical removal, and chemotherapy.
However, difficulties in surgical excision, and the severe adverse
effects associated with irradiation and chemotherapy, hinder these
approaches (4,5). Furthermore, the advanced chemotherapy
regimen has been reported as having limited or no impact on brain
tumors due to the poor penetration profile of the drug through the
highly variable, heterogeneous blood brain barrier (BBB) (6). In addition, a protective blood-brain
tumor-barrier (BBTB) has been shown to form around tumor cells
alongside the progression of GBM, which regulates the entry of
small molecules (7). Therefore,
any approach that can overcome the tight extracellular junctions of
both the BBB and BBTB would be a promising treatment solution.
Nanotechnology may be a prospective method to
overcome the BBB and BBTB. Polymeric nanoparticles (NP), or
polymeric micelles, have drawn significant attention due to their
ability to surpass the physiological barriers and improve the
delivery of therapeutic agents into the brain (8,9). The
enhanced permeability and retention effect is the main factor that
allows the preferential accumulation of NP in the tumor
fenestrations. However, limited penetrability, and non-uniform
distribution into other body parts, impairs the performance of
simple carrier-mediated delivery systems (10). Receptor-mediated active targeting
moiety-tagged NPs may be a feasible approach in the translocation
of the carriers. Cyclic Arginine-Glycine-Aspartic acid (cRGD) has
been selected as a targeting moiety to promote the biological
interactions between the delivery carrier and the overexpressed
receptor in the tumor cells (11).
Specifically, αvβ3 and αvβ5 integrins, which are over-expressed in
tumor endothelial and GBM cells, have been shown to have a
selective affinity to cRGD (12).
Chen et al (13)
successfully demonstrated the in vivo imaging of GBM, with
the aid of an RGD-containing probe. Furthermore, an RGD-iron oxide
NP conjugate has been developed, and shown to enhance the targeting
efficiency in brain tumors (14).
Docetaxel (DTX) is an anti-mitotic taxane drug,
considered to be one of the most effective drugs against brain
tumors (15). The present study
developed polylactic acid-polyethylene glycol (PLA-PEG)-based long
circulating, DTX-loaded, cyclic RGD-attached polymeric micelles
[DTX-PLA-PEG/c(RGDyK)]. These micelles could prolong the blood
circulation time of the carriers, and specifically target the
integrin receptors known to be over-expressed in brain tumors. The
PEG provided an anti-fouling property and extended the circulation
time of the carriers, whilst the cRGD specifically targeted the
tumor cells. The aim of the present study was to develop a
functional NP system which could be transported across the
physiological barriers of the brain, and effectively deliver the
therapeutic load. Pharmacokinetic and biodistribution studies were
performed to quantify the amount of drug accumulated in the brain
tissue. The antitumor efficacy of the system was determined using a
U87MG bearing xenograft tumor model, by measuring the tumor volume
and survival rate index. The study investigated whether cRGD-linked
polymeric micelles can effectively penetrate the brain tumor and
exhibit a potent antitumor effect.
Materials and methods
Materials
The materials used for the subsequent experiements
were provided by the following suppliers. The methoxyl
poly(ethylene glycol) (mPEG-OH, 2 kDa) and the
maleimide-poly(ethylene glycol) (mal-PEG-OH, 3.5 kDa) were obtained
from Sigma-Aldrich (Hong Kong, China). The
benzotriazole-N,N,N,N′-tetramethyl-uronium-hexafluorophosphate was
from American Bioanalytical Inc. (Natick, MA, USA).
Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
was obtained from GL Biochem Ltd (Shanghai, China).
Diisopropylethylamine and H-Gly-2-Chlorotrityl resin were both
supplied by Fluka (Sigma-Aldrich). 3,3′-dithiodipronic acid and
D,L-dithiotheriol (DTT) were purchased from Aladdin Reagents Co.
(Shanghai, China). Cyclo[RGDfK(CX-)] (cRGD peptide,
X=6-aminocaproic acid:-Acp) was purchased from Peptide Institute
Inc. (Osaka, Japan). DTX was procured from Sigma-Aldrich
(China).
Synthesis of RGD-PLA-PEG polymer
The synthesis of PLA-PEG was performed as reported
by previous methods (16). The
PLA-PEG was conjugated with the thiolated c(RGDyK) to form
c(RGDyK)-PLA-PEG as previously reported (17). Briefly, 3,3′-dithiodipronic acid
was activated by dicyclohexylcarbodiimide (DCC) and
N-hydroxysuccinimide (NHS) and the resulting active ester was
reacted with c(RGDyK). The obtained product was reduced with DTT to
obtain the thiolated cRGD peptide. To conjugate the peptide with
PLA-PEG, mal-PLA-PEG was dissolved in acetonitrile (ACN) and a
thin-film was formed by rotary evaporation. Phosphate-buffered
saline (PBS-hydrated PLA-PEG was added to the c(RGDyK)-SH and
stirred overnight. The resulting product was extracted and
characterized by nuclear magnetic resonance. The polymers were
characterized using 1H nuclear magnetic resonance (NMR;
solvents, CDCl3, D2O and deuterated-dimethyl
sulfoxide (DMSO); temperature, 25°C). The NMR spectra were recorded
using a JEOL Alpha 500 spectrometer (500 MHz; Jeol, Ltd., Tokyo,
Japan).
Preparation of DTX-loaded micelles
The DTX-loaded polymeric micelles were formed using
a thin-film hydration technique. Briefly, 10 mg of PLA-PEG and 5 mg
of DTX were dissolved in 5 ml of ACN. The mixture was
rotary-evaporated and the thin-film was subsequently hydrated with
PBS. For the c(RGDyK)-tagged micelles, 5% w/w c(RGDyK)-PLA-PEG was
mixed with 95% DTX-PLA-PEG. The resulting micelles were evaluated
for drug loading and dynamic light scattering (DLS) characteristics
using a Malvern Zetasizer (Malvern Instruments Ltd., Malvern,
UK).
Particle size distribution and zeta
potential
The micellar solutions were suitably diluted, in
order to analyze the particle size distribution and zeta potential,
using the DLS method. A Malvern Zetasizer was used to determine the
DLS characteristics. All measurements were performed at a fixed
angle of 90° at 25°C. The results were expressed as the size ±
standard deviation.
DTX loading efficiency
The loading efficiency of the carrier was calculated
from the total amount of drug added, versus the amount of drug
entrapped within the nanoparticles. Briefly, DTX-PLA-PEG (DPP), and
RGD/DTX-PLA-PEG (RDPP) were filtered using an Amicon®
centrifugal filter (Millipore, Billerica, MA, USA) at 550 × g for
10 min. The filtrate was then analyzed for unentrapped drugs by
high performance liquid chromatography (HPLC). The mobile phase
(acetonitrile : water, 60:40, pH 3.5) was set at 1 ml/min with an
absorbance of 254 nm.
In vitro release study
The in vitro release of DTX from both PLA-PEG
and RGD/PLA-PEG was evaluated by dialysis. A total of 1 ml of NP
dispersions were placed in a dialysis bag (3000 MW cutoff;
Spectra/Por®, Spectrum Laboratories, Inc., Rancho
Dominguez, CA, USA) and both the borders were sealed with a
dialysis clip. The dialysis bag was incubated in 30 ml release
media (PBS, pH 7.4), containing 1% Tween® 80 as a
solubilizer. The bag was placed in an automated shaker maintained
at 2 × g and 37°C. At a specified time (1, 2, 4, 8, 12, 24, 48, 72,
96, 120, 144 and 168 h), the release media was collected and
replaced with an equal amount of fresh media. The amount of
released drug was quantified by HPLC.
Cytotoxicity assay
U87MG human malignant glioma and 9L rat gliosarcoma
cells (ATCC, Manassas, VA, USA) were cultured in normal RPMI-1640
media supplemented with 10% fetal bovine serum and 1%
penicillin-streptomycin in a 5% CO2 and 95% humidified
atmosphere. The viability of the cells was determined by MTT assay
(Sigma-Aldrich). Briefly, the cells were seeded into a 96-well
plate, at a seeding density of 1×104 cells, and
incubated for 24 h. The following day, the media was refreshed and
various concentrations of free DTX, DPP or RDPP were exposed to the
cells followed by 24 and 48 h incubations. The cells were then
washed and treated with MTT solution (5 mg/ml in serum-free media)
and incubated for 3 h. The purple blue formazan crystals were
extracted by the addition of DMSO and the absorbance was detected
using a plate reader (Multiskan Ascent, Labsystems SA,
Cergy-Pontoise, France) at 570 nm.
Cellular uptake study
A cellular uptake study was performed to investigate
the kinetics of drug internalization. A total of 1×106
U87MG and 9L cells were seeded in a 6-well plate and allowed to
adhere for 24 h. The following formulations, free DTX, DPP or RDPP
(DTX equivalent 20 μg/ml) were incubated with the cells for 1–4 h.
Following the incubation, the media was removed and the cells were
washed twice with PBS. An aliquot of lysis buffer was added to lyse
the cells, followed by centrifugation at 500 × g for 10 min. The
lysis buffer (Sigma-Aldrich) consisted of 50 mmol/l Tris, 150
mmol/l NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium
dodecyl sulfate, 1 mmol/l sodium orthovanadate, 1 mmol/l sodium
fluoride, 1 mmol/l EDTA and 1 mmol/l phenylmethylsulfonyl fluoride.
The supernatant was collected and analyzed by HPLC. The percentage
of cellular uptake of DTX from the various formulations was
estimated by normalizing to the initial DTX concentration
administered.
Cell cycle analysis
U87MG and 9L cells were seeded in 6-well plates at a
density of 3×105 cells/well with RPMI-1640 media for 24
h. The cells were then exposed to various doses of free DTX, DPP or
RDPP and further incubated for 24 h. Following incubation the cells
were washed and further incubated with propidium iodide (20 μg/ml)
for 30 min The DNA content was measured for 10,000 events for each
sample by flow cytometry using a BD FACS Atira II (BD Biosciences,
Franklin Lakes, NJ, USA). The data were plotted using Cell Quest™
(BD Biosciences) software.
Analysis of glioma spheroids in
vitro
The inhibitory effect of various formulations on the
growth of glioma spheroids was investigated. U87MG tumor spheroids
were incubated for a period of seven days, after which the media
was refreshed, prior to the application of the formulations. The
glioma cells, incubated in RPMI-1640 media, were exposed to free
DTX, DPP, or RDPP. On days 0, 2, 4, 6, and 8, the tumor spheroids
were observed under an inverted microscope. The inhibitory effects
of the various formulations were calculated using the following
formula: V=(π × dmax × dmin)/6, where
dmax and dmin refer to major and minor
diameters, respectively. The spheroid tumor volume at day 8 was
calculated using the following formula: ratio
%=(Vday8/Vday0) × 100. Vday0 is
the spheroid volume t day 0. The sections were observed using a
Zeiss Axioscope fluorescence microscope and photographed using a
digital AxioCam mRM camera (Zeiss, Oberkochen, Germany).
Antitumor efficacy of
c(RGDyK)-PLA-PEG-DTX
Nude mice were obtained from the Experimental Animal
Center of Soochow University (Suzhou, China) and maintained under
regular light and dark conditions with free access to food. The
study was approved by the Institutional Animal Care Committee of
Soochow University. U87MG tumor bearing xenograft nude mice were
developed by subcutaneously injecting 1×106 cells into
the right flank of the mice. The tumor growth was monitored until
it reached ~100–150 mm3. Approximately two weeks after
the tumor cell injection, the experiment was started. The mice were
randomly divided into five groups (n=6/group). Groups I to IV were
administered saline, free DTX, DPP or RDPP, and group V was
identified as the untreated, control group. A total of 100 μl of
each formulation (10 mg/kg body weight of DTX) was injected into
the mice subcutaneously twice a week for two weeks. The tumor size
was measured biweekly using a caliper, and the tumor volume (V) was
calculated using the following formula V=1/2[L ×( W)2];
where L= length and W= width.
Pharmacokinetics and biodistribution
The pharmacokinetics study was performed in seven
week old mice (~20 g). The mice were divided into three groups
(n=5/group). Each group was administered with free DTX, DPP or RDPP
(10 mg/kg DTX) via tail vein injection. The blood samples were
collected at 0.25, 0.5, 1, 2, 4, 6, 8, 12, and 24 h. The samples
were immediately centrifuged at 800 × g for 15 min and stored at
−20°C until further analysis. To prepare the plasma samples, 100 μl
of methanol containing 50 ng/ml of DTX was added to 50 μl of
plasma. The mixture was vortexed for 30 min and subsequently
centrifuged. The supernatant was collected and analyzed for DTX
content by HPLC.
For the biodistribution study, brain tumor-bearing
mice were developed as previously described in the above methods.
Three weeks after the tumor implantation, the mice were divided
into three groups (n=14/group). The groups were administered free
DTX, DPP or RDPP (10 mg/kg DTX) via tail vein injection. At a
specified time point, two mice were sacrificed and their organs
were surgically removed and preserved on ice. At 0.5, 1, 2, 4, 6,
12 and 24 h, mice were sacrificed by carbon dioxide inhalation in a
closed tube, according to the ethical guidelines. The organs were
macerated using a high pressure homogenizer (10,000 × g;
Ultra-Turrax T25 Homogenizer; IKA®-Werke GmbH & Co.
KG, Staufen, Germany) and the drug levels present in the homogenate
of each organ were analyzed by HPLC.
Statistical analysis
Statistical significance was determined using
one-way analysis of variance, followed by Tukey’s post hoc test.
Statistical significance was evaluated using SPSS version 5 (SPSS,
Inc., Chicago, IL, USA). All values are expressed as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results and Discussion
Characterization of DPP and RDPP
micelles
Brain tumors are one of the most aggressive and
difficult tumors to treat, partly due to the complexity of the
anatomical location. Furthermore, both the BBB and BBTB hamper any
therapeutic strategy made to increase the drug accumulation in the
brain. An attractive prospect of brain chemotherapy is the use of
targeted drug delivery, which target cellular receptors in the
brain. Such cellular responsive brain targeting systems are
expected to increase the drug concentration in brain tumors. In the
present study RGD, which has a high affinity for the αvβ3 and αvβ5
integrins in GBM cells, was conjugated with PLA-PEG by a DCC/NHS
reaction. DTX was encapsulated within PLA-PEG and RGD/PLA-PEG,
resulting in the formation of DPP and RDPP micelles respectively.
The mean particle size of DPP was ~90 nm with a uniform size
distribution (polydispersity index ~0.150) (Table 1). RGD-linking to the PLA-PEG
slightly increased the size of micelles, however the size remained
small (~120 nm) with a narrow distribution of particles (Fig. 1A). The RGD linking was evident as
the overall zeta potential of micelles decreased from ~-28 to ~-22
mV (Fig. 1B). Both of the micelles
showed high entrapment efficiency of >90% and a markedly high
loading capacity of ~30%. RGD substitution did not decrease the
drug loading capacity of the micelles and presented self-assembled
mono-dispersed spherical shaped particles.
 | Table IPhysicochemical characterization of
micelles. |
Table I
Physicochemical characterization of
micelles.
| Micelle | Size (nm) | PDI | Charge (m) | EE (%) | DLS (%) |
|---|
| DTX-PLA-PEG | 90.5±2.5 | 0.158±0.002 | −28.6±1.23 | 94.2±2.6 | 30.26±3.42 |
|
DTX-PLA-PEG/c(RGDyK) | 118.4±1.6 | 0.234±0.003 | −22.4±1.34 | 90.4±3.6 | 28.65±1.46 |
In vitro drug release
The cumulative DTX release from both the DPP and
RDPP micelles are presented in Fig.
1C. Free DTX exhibited a burst release profile with 60% of drug
released within 24 h. The drug-loaded micelles however,
significantly controlled the release rate without any burst-release
phenomenon (P<0.05). As expected, DPP and RDPP micelles showed
similar release profiles throughout the study period. During a 24 h
incubation <20% of DTX was released, whilst ~60% of the drug had
been released by the end of seven days. Such a sustained release
profile would be of significant importance for systemic delivery,
or brain tumor targeting, where a substantial amount of drug is
expected to locate in the intracellular environment. To investigate
the release kinetics, the data were fitted to various mathematical
models including zero order, first order and Higuchi. It was found
that the micellar formulations were best fitted to the Higuchi
model. These results indicated that diffusion was the governing
mechanism of release. In addition, the Korsemeyer-Peppas equation
indicated the presence of more than one mechanism of release
(18).
Cytotoxicity assay
The cytotoxicity profiles of free DTX, DPP and RDPP
when exposed to U87MG and 9L brain cancer cell lines for 24 and 48
h are shown in Fig. 2. All of the
formulations showed time and concentration-dependent cytotoxicity
on both of the cell lines. Marked differences were observed in the
RDPP group, which exhibited significantly greater cytotoxicity as
compared with the free drug and DPP groups (P<0.05). It was
hypothesized that the pronounced cytotoxicity of RDPP may result
from the enhanced internalization of the particles. The potent
cytotoxic action of RGD-linked particles may increase the chances
of brain cancer recovery. Conversely, free DTX showed higher
cytotoxicity as compared with DPP, which is consistent with a
previously published report which stated that free drugs diffuse
easily into the cell nucleus, whereas micellar drugs have to be
detached from the NP prior to exhibiting any therapeutic action
(19). The 9L cells were
relatively more sensitive to all of the formulations, as compared
with the U87MG cells. The difference in the cytotoxic effects on
both of the cell lines may be attributed to the difference in
genetic origin and indigenous biological behavior (20). In order to precisely quantify the
effects of the individual therapeutic systems, the half maximal
inhibitory concentration (IC50) was evaluated. The
IC50 values of free DTX, DPP and RDPP in U87MG were
8.56, 9.89, and 3.26 μg/ml, respectively following 24 h incubation,
while these values decreased to 2.18, 2.38, and 1.05 μg/ml
following 48 h incubation. The respective values in 9L cells were
6.85, 7.92, and 2.54 μg/ml for the 24 h incubation and 1.68, 1.74,
and 0.76 μg/ml for the 48 h incubation periods. These results
suggest that the specific interactions of the targeting ligand and
the enhanced cellular uptake of carriers during a longer incubation
period may greatly increase the drug cytotoxicity. Therefore,
actively targeted RDPP may be preferentially taken up by the cells
through receptor-mediated endocytosis resulting in increased
cytotoxicity, whereas non-targeted micelles may enter by normal
passive uptake mechanisms only (21).
Cell uptake
The majority of delivery systems exert their
therapeutic action following internalization into the cells.
Therefore, the present study investigated the cellular uptake
efficiency of U87MG and 9L cancer cells, that can augment the
accumulation of drugs. The cellular uptake of both the free drug
and the NPs increased in a time-dependent manner up to 4 h
(Fig. 3a,b). Maximum
internalization was completed by 1 h, followed by a relatively
slower uptake until the end of 4 h. As expected, the RDPP group
exhibited a significantly higher uptake, as compared with that of
either the free drug or DPP groups (P<0.05). This may be due to
the presence of the RGD moiety on the surface, which has a
selective affinity towards the overexpressed αvβ3 and αvβ5
integrins on the cancer cell surface (22). It has previously been demonstrated
that RGD-based complexes are internalized by a combination of
various mechanisms; including clathrin and caveolae-mediated
endocytosis, clathrin and caveolae-independent endocytosis and
macropinocytosis (23,24). The free DTX was continuously
internalized in both of the cell lines, through passive diffusion.
Specifically, 9L GBM cells showed relatively higher cellular uptake
as compared with the U87MG cells. These data suggest that the RGD
delivery system may be preferentially recognized by the integrin
receptors present on the GBM cell surface, and may be internalized
by various receptor and energy-dependent processes. Consequently,
the active targeting moiety may be used to target the cancer cells
upon intravenous injection and the higher uptake efficiency can
rapidly remove the drug from the circulation.
Cell cycle analysis
The polymerization and depolymerization of
microtubules has an important role in biological processes. DTX
tightly binds microtubules, and promotes their stabilization,
resulting in the mitotic arrest of cancer cells (25). It is well known that DTX induces
cell cycle arrest through the impairment of mitosis and chromosomal
damage, thereby inducing typical G2/M phase arrest (26). The cell cycle analyses of the cells
treated with the different formulations are presented in Fig. 3C and D. The data shows the presence
of the cell populations in the different phases upon treatment with
the drug formulations. In the control group ~50% cells were in the
G1 phase, whereas only ~7% cells were in the G2/M phase. In the
RDPP group ~45% cells were shown to be in G2/M phase, as compared
with ~30 and ~20% for the DPP and free DTX groups in U87MG cells,
respectively. A similar trend was observed in 9L cells, however the
proportion of cells in G2/M phase was relatively higher as compared
with U87MG cells. The superior cell arresting effects of the
RDK-linked formulations was consistent with the higher cellular
uptake and cytotoxic potential, demonstrated in both of the cancer
cell lines. It was hypothesized that RDPP may promote the
stabilization of microtubules and differentiation of cancer cells.
This may result in the downregulation of tubulin, leading to the
impairment of mitotic spindle formation and eventually cell cycle
arrest.
Analysis of in vitro spheroid
inhibition
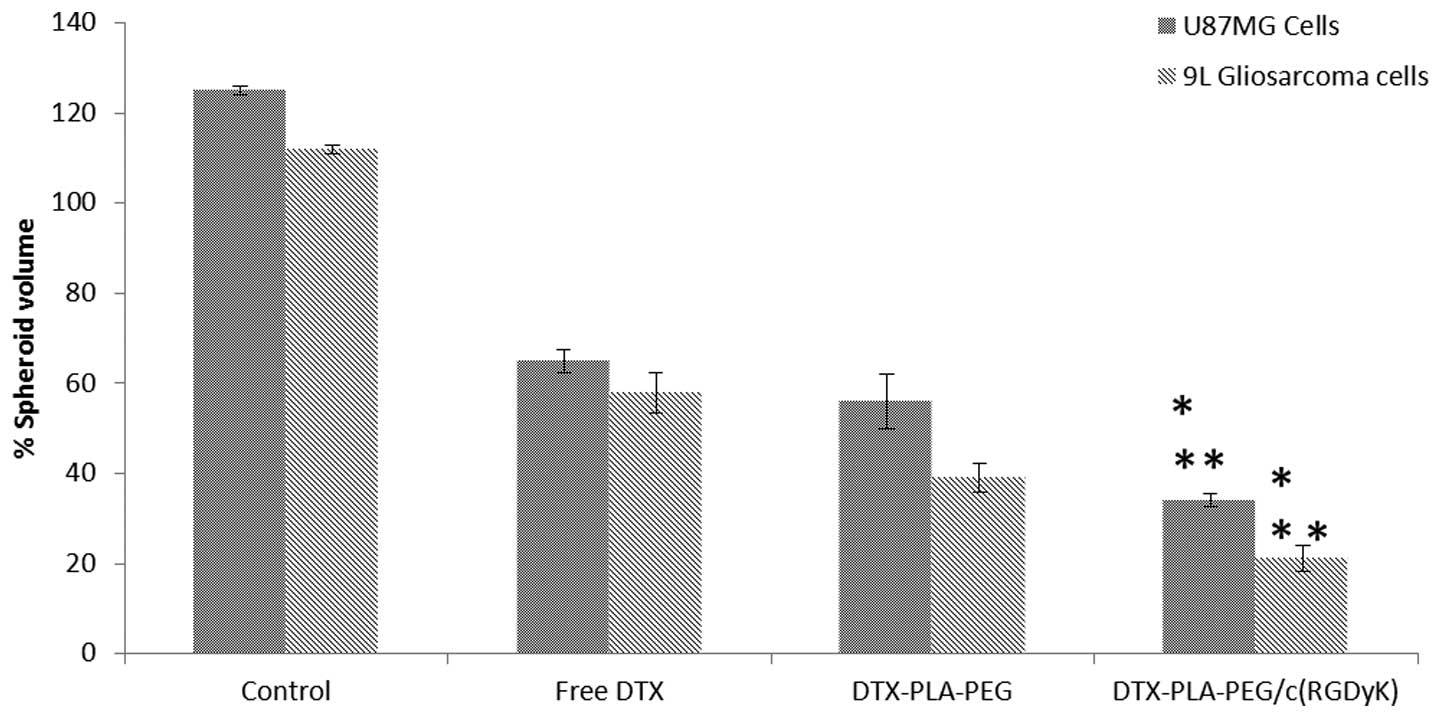
The inhibitory effects of free DTX, DPP and RDPP on
the growth of U87MG and 9L spheroids are presented in Fig. 4. Spheroids in the untreated group
grew rapidly and attained a maximum volume of ~125 and ~110% for
U87MG and 9L cells, respectively. In contrast, the free and
micellar drugs significantly inhibited the growth of the spheroids
(P<0.05). The spheroids appeared distorted, disintegrated,
shrunken and had lost their typical three dimensional structures.
RDPP treatment resulted in a >80% reduction in spheroidal
volume, indicating its superior inhibitory effects. Free DTX could
directly kill the cells on the surface, however transport across
the spheroid proved difficult. In contrast, hydrophobic
core-containing micelles exhibited a preferential uptake.
RGD-mediated cellular uptake may further increase the drug
concentration within the spheroids. The inhibitory effect was in
the order of RDPP > DPP > DTX. The stronger inhibitory
effects of RDPP may be due to its better permeability within the
cells, resulting in superior in vitro therapeutic effects
(27).
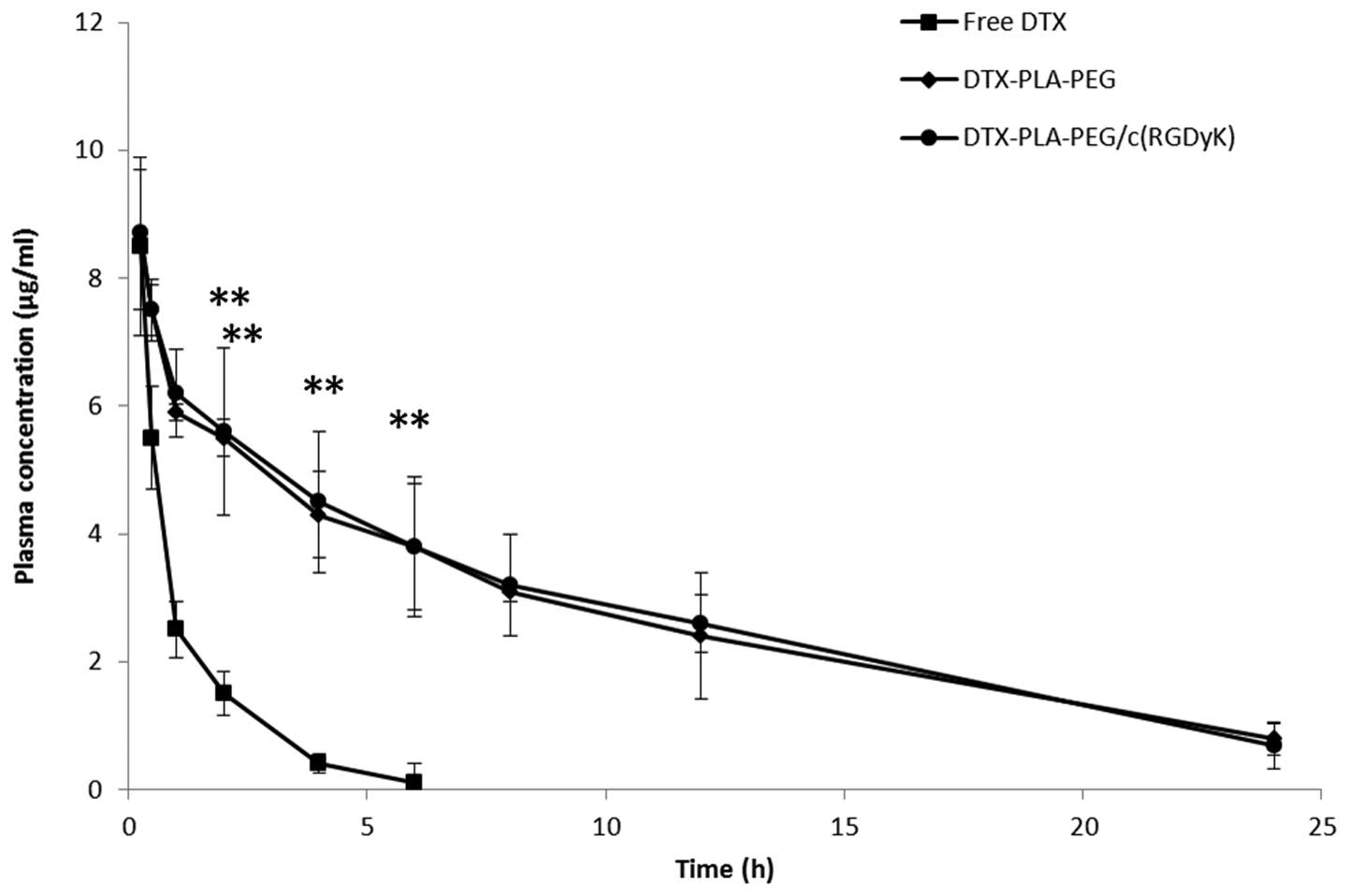
Evaluation of pharmacokinetics
The plasma concentration-time profiles of DTX,
following a single dose intravenous administration of free DTX, DPP
or RDPP, are presented in Fig. 5.
Free DTX was immediately removed from the blood circulation and
reached the detection limit within 6 h of the study period. In
contrast, the micellar formulations significantly enhanced the
circulatory performance of DTX (P<0.05). Substitution of PEG
with the RGD block did not hamper the long circulation profile of
RDPP, and a substantial concentration of DTX was detected
throughout the study period (24 h). RDPP showed a 3–4 fold higher
area under curve (36.85±4.52 h μg/ml) as compared with the free DTX
(7.96±2.62 h μg/ml). Consequently, RDPP showed a significantly
higher terminal half-life of 4.57 h, as compared with 0.91 h of the
free drug. The longer circulation profile of RDPP may promote the
likelihood of BBB permeation and higher tumor targeting
ability.
Biodistribution study
The concentration of DTX present in the mouse
organs, including the brain, heart, spleen, liver, kidney, and
lungs were investigated at different time points. The study was
performed following a single dose intravenous injection of each
formulation via the tail vein. The results clearly showed that RDPP
was able to deliver the DTX into the brain at a significantly
higher level as compared with the non-targeted DPP micelles and the
free drug (P<0.05; Fig. 6A).
RDPP exhibited ~1 μg DTX/g tissue after 30 min; this level
gradually decreased up to 6 h, after which no drug was detected.
Conversely, DPP showed ~0.5 μg DTX/g tissue at 30 min, which
immediately decreased to the lowest level at 1 h, after which no
drug was detected. No drug was detected in the free DTX group. The
significantly higher concentration (P<0.05) observed in the RDPP
group was consistent with its ability to interact with the cancer
cells via receptor mediated endocytosis, whereas DPP could not
influence the cellular uptake beyond a certain limit. Furthermore,
the BBB is randomly disrupted in brain tumors, along with the
components of the tumor blood vessels including the basement
membrane, pericytes and the endothelial cells which provide entry
for the nanosized particles (28).
This further proves that passive targeting is inefficient in
maintaining adequate concentrations of drugs in brain cancer cells.
RGD-linking, however, along with an increased circulative ability,
markedly enhanced the drug concentration levels. In the heart,
liver and spleen, RDPP accumulated at relatively low levels,
suggesting that drug-related systemic cytotoxicity may be
effectively avoided (Fig. 6B–F).
The reduced presence of the drug in the lung, in response to
treatment with either of the micellar formulations, may be due to
its nanosize, which may avoid enhanced uptake by the lung
tissue.
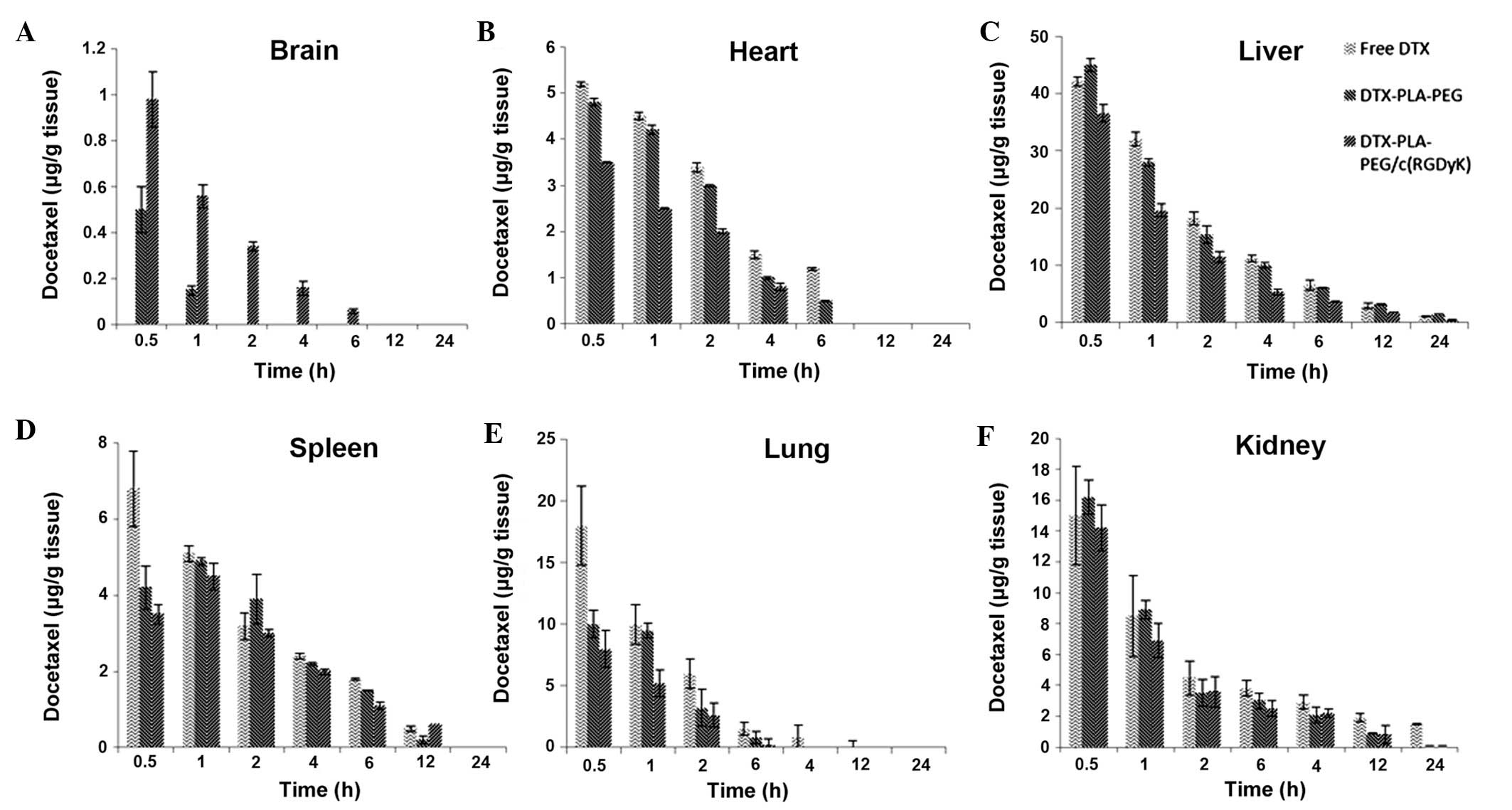 | Figure 6Biodistribution of free docetaxel
(DTX), docetaxel polylactic acid-polyethylene glycol (DTX-PLA-PEG)
or DTX-PLA-PEG/cyclic (Arginine-Glycine-Aspartic
acid-D-Tyrosine-Lysine) [DTX-PLA-PEG/c(RGDyK)] micelles in U87MG
tumor-bearing nude xenograft mice in different organs following
intravenous administration of the different formulations. (A)
Brain, (B) heart, (C) liver, (D) spleen, (E) lung, (F) kidney. The
mice were sacrificed at 0.5, 1, 2, 4, 6, 12, and 24 h
post-injection. |
Antitumor efficacy
A U87MG-bearing tumor model was developed and
treated with the various formulations. As shown in Fig. 7, RDPP significantly inhibited the
growth of glioblastoma tumors, as compared with that of the control
and free drug treated groups of mice (P<0.05). The tumor volume
remained the same until day four, however after that, significant
differences were observed between the different groups. Tumors in
the saline and control groups grew rapidly and attained maximum
size. The marked antitumor efficacy of the RGD-linked micelles was
attributed to the specific binding with the highly angiogenic
neovasculature of subcutaneously grown GBM cells. Consistent with
the long circulation profile and the drug accumulation in the
specific tumors, enhanced tumor regression may be anticipated.
Similar results have been reported by previous studies which
demonstrated the improved antitumor effects for RGD-linked delivery
systems (29,30).
The body weight fluctuations of the mice were
assessed in terms of in vivo systemic toxicity of the drug
and delivery system. Body weight of the individual mice was noted
throughout the study period alongside the tumor volume measurement.
The mice administered with saline and the control group experienced
a gain in body weight, attributed to an increased tumor volume. In
the DPP and RDPP administered groups, no loss of body weight was
observed, indicating a lack of any serious drug-related
toxicity.
Thus far, glioblastoma, which represents one of the
deadliest forms of cancer, is unresponsive to chemotherapy mainly
due to the poor penetrability of therapeutics across the BBB and
BBTB. Therefore, the aim of the present study was to design a
delivery system, appended to a targeting ligand, that could
overcome these physiological barriers and deliver the anticancer
drugs into the glioma cells. GBM is generally characterized by
angiogenesis and over-expression of the integrin αvβ3 receptor on
the surface of the tumor neovasculature. In the present study
c(RGDyK) peptide was selected as a specific targeting agent to the
integrin receptors. The results clearly showed that RDPP could
effectively bind the αvβ3 receptor, facilitating the cellular
uptake and inreasing the cytotoxicity of the drug. Overexpression
of the αvβ3 receptor resulted in an enhanced accumulation of RDPP
in the brain tumor cells in vivo, whilst non-targeted
micelles had limited access. RGD-linked micelles showed pronounced
tumor regression with an excellent safety profile. Therefore, the
results clearly revealed the superior antitumor efficacy of
c(RGDyK)/DTX-PLA-PEG in an experimental brain tumor model.
In conclusion, DPP and RDPP were successfully
prepared and subjected to various in vitro and in
vivo characterizations. RDPP showed enhanced accumulation in
U87MG and 9L glioblastoma cell lines, through various endocytic
pathways, and displayed pronounced cytotoxic effects in the cell
lines with markedly lower IC50 values. Furthermore, RDPP
showed a typical G2/M phase arrest and stronger microtubule
stabilizing effects. Consequently, high accumulation and
distribution of RDPP resulted in tremendous growth inhibition of
glioma spheroids, as compared with the non-targeted DPP and free
drug. The ability of RDPP to bind the αvβ3 integrin receptor
resulted in a high accumulation of DTX in the brain glioblastoma
cells, which in turn may be due to the specific interaction of
micelles, small particle size and an extended blood circulation
profile. The enhanced tumor regression in the U87MG tumor-bearing
mice further confirmed the superior anti-glioblastoma efficacy of
RDPP. The findings of the present study suggest that c(RGDyK)
linked to delivery carriers may be a potential strategy for the
treatment of advanced brain cancer.
Acknowledgements
The authors would like to thank Dr. Shiam for proof
reading the manuscript.
References
|
1
|
Jain RK, di Tomaso E, Duda DG, et al:
Angiogenesis in brain tumors. Nat Rev Neurosci. 8:610–622. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ong BY, Ranganath SH, Lee LY, et al:
Paclitaxel delivery from PLGA foams for controlled release in
post-surgical chemotherapy against glioblastoma multiforme.
Biomaterials. 30:3189–3196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant Gliomas in
Adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jones TS and Holland EC: Standard of care
therapy for malignant glioma and its effect on tumor and stromal
cells. Oncogene. 31:1995–2006. 2012. View Article : Google Scholar
|
|
5
|
Rich JN and Bigner DD: Development of
novel targeted therapies in the treatment of malignant glioma. Nat
Rev Drug Discov. 3:430–446. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ningaraj NS: Drug delivery to brain
tumours: challenges and progress. Expert Opin Drug Deliv.
3:499–509. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sarin H: Recent progress towards
development of effective systemic chemotherapy for the treatment of
malignant brain tumors. J Transl Med. 7:772009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xin H, Sha X, Jiang X, Zhang W, Chen L and
Fang X: Anti-glioblastoma efficacy and safety of paclitaxel-loading
Angiopep-conjugated dual targeting PEG-PCL nanoparticles.
Biomaterials. 33:8167–8176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gu G, Xia H, Hu Q, et al: PEG-co-PCL
nanoparticles modified with MMP-2/9 activatable low molecular
weight protamine for enhanced targeted glioblastoma therapy.
Biomaterials. 34:196–208. 2013. View Article : Google Scholar
|
|
10
|
Peer D, Karp JM, Hong S, Farokhzad OC,
Margalit R and Langer R: Nanocarriers as an emerging platform for
cancer therapy. Nat Nanotechnol. 2:751–760. 2007. View Article : Google Scholar
|
|
11
|
Danhier F, Le Breton AL and Préat V:
RGD-based strategies to target alpha(v) beta(3) integrin in cancer
therapy and diagnosis. Mol Pharm. 9:2961–2973. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schottelius M, Laufer B, Kessler H and
Wester HJ: Ligands for mapping alphavbeta3-integrin expression in
vivo. Acc Chem Res. 42:969–980. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen X, Park R, Shahinian AH, et al:
18F-labeled RGD peptide: initial evaluation for imaging brain tumor
angiogenesis. Nucl Med Biol. 31:179–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Hou Y, Tohme M, et al: Pegylated
Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor
alphavbeta3-integrin expression. J Nucl Med. 45:1776–1783.
2004.PubMed/NCBI
|
|
15
|
Gajbhiye V and Jain NK: The treatment of
Glioblastoma Xenografts by surfactant conjugated dendritic
nanoconjugates. Biomaterials. 32:6213–6225. 2011.PubMed/NCBI
|
|
16
|
Nasongkla N, Bey E, Ren J, et al:
Multifunctional polymeric micelles as cancer-targeted,
MRI-ultrasensitive drug delivery systems. Nano Lett. 6:2427–2430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhan C, Gu B, Xie C, Li J, Liu Y and Lu W:
Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid)
micelle enhances paclitaxel anti-glioblastoma effect. J Control
Release. 143:13620–142. 2010. View Article : Google Scholar
|
|
18
|
Ramasamy T, Khandasami US, Ruttala H and
Shanmugam S: Development of solid lipid nanoparticles enriched
hydrogels for topical delivery of anti-fungal agent. Macromol Res.
20:682–692. 2012. View Article : Google Scholar
|
|
19
|
Carrion C, de Madariaga MA and Domingo JC:
In vitro cytotoxic study of immunoliposomal doxorubicin targeted to
human CD34(+) leukemic cells. Life Sci. 75:313–328. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carvalho JC, Perazzo FF, Machado L and
Bereau D: Biologic activity and biotechnological development of
natural products. BioMed Res Int. 2013:9717452013. View Article : Google Scholar
|
|
21
|
Gupta B and Torchilin VP: Monoclonal
antibody 2C5-modified doxorubicin-loaded liposomes with
significantly enhanced therapeutic activity against intracranial
human brain U-87 MG tumor xenografts in nude mice. Cancer Immunol
Immunother. 56:1215–1223. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu J and Shapiro JI: Endocytosis and
signal transduction: basic science update. Biol Res Nurs.
5:117–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang X, Sha X, Xin H, et al:
Self-aggregated pegylated poly (trimethylene carbonate)
nanoparticles decorated with c(RGDyK) peptide for targeted
paclitaxel delivery to integrin-rich tumors. Biomaterials.
32:9457–9469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang X, Sha X, Xin H, et al:
Integrin-facilitated transcytosis for enhanced penetration of
advanced gliomas by poly(trimethylene carbonate)-based
nanoparticles encapsulating paclitaxel. Biomaterials. 34:2969–2979.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Downing KH and Nogales E: Tubulin and
microtubule structure. Curr Opin Cell Biol. 10:16–22. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miller ML and Ojima I: Chemistry and
chemical biology of taxane anticancer agents. Chem Rec. 1:195–211.
2001. View
Article : Google Scholar
|
|
27
|
Gao H, Yang Z, Zhang S, et al:
Glioma-homing peptide with a cell-penetrating effect for targeting
delivery with enhanced glioma localization, penetration and
suppression of glioma growth. J Control Release. 172:921–928. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kanazawa T, Taki H, Tanaka K, Takashima Y
and Okada H: Cell-penetrating peptide-modified block copolymer
micelles promote direct brain delivery via intranasal
administration. Pharm Res. 28:2130–2139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao H, Wang JC, Sun QS, et al: RGD-based
strategies for improving antitumor activity of paclitaxel-loaded
liposomes in nude mice xenografted with human ovarian cancer. J
Drug Target. 17:10–18. 2009. View Article : Google Scholar
|
|
30
|
Saad M, Garbuzenko OB, Ber E, et al:
Receptor targeted polymers, dendrimers, liposomes: which
nanocarrier is the most efficient for tumor-specific treatment and
imaging? J Control Release. 130:107–114. 2008. View Article : Google Scholar : PubMed/NCBI
|