1. Introduction
Postoperative pulmonary dysfunction is one of the
most common complications associated with cardiopulmonary bypass
(CPB) and it is particularly associated with an increased morbidity
and mortality in high-risk patients having repeat coronary artery
bypass surgery (1). The clinical
presentation of CPB-induced lung injury varies from mild dyspnea to
fully developed adult respiratory distress syndrome in ~2% of cases
undergoing CPB, which, in itself, results in a 50% mortality rate
(2,3). The damaged lung following CPB is
characterized by abnormal functional, physiological, biochemical
and histological alterations, including abnormal gas exchange and
poor lung mechanics, increased lung permeability and pulmonary
vascular resistance, the presence of neutrophil elastase and high
expression of matrix metalloproteinases (MMPs) as well as alveolar
edema and extravasation of erythrocytes and neutrophils (4).
Although impairment of lung function can occur
following any major surgery, CPB appears to cause additional damage
to the lung compared with off-pump coronary artery bypass grafting
(5). It has been well recognized
that the contact of blood cells with the artificial surface of the
CPB circuit can activate polymorphonuclear cells (PMNs) and PMN
activity can be further enhanced by proinflammatory factors
(6), suggesting increased
inflammation associated with CPB. Furthermore, ischemia-reperfusion
also significantly contributes to CPB-induced inflammation.
Diegeler et al (7) observed
a significantly increased release of activated complement factors
C5a and C3d, interleukin (IL)-8 and IL-10 and prolonged elevation
of tumor necrosis factor-α (TNF-α) receptors p55 and p75 in
patients undergoing CPB compared with the off-pump groups. In
addition, postoperative white blood cell, neutrophil and monocyte
counts and oxidative stress are also significantly higher in the
on-pump than the off-pump group (8,9).
Thus, CPB appears to be able to elicit or exasperate a series of
inflammatory reactions, ultimately leading to lung injury. The
purpose of this review was to discuss the therapeutic strategies
for reducing CPB-induced pulmonary inflammation with a particular
focus on the intervention approaches targeting the proinflammatory
cytokine TNF-α.
2. The underlying mechanism of
TNF-α-mediated lung injury during CPB
Effects of CPB on TNF-α level in the
serum and lung tissue
Numerous studies on clinical CPB and animal models
of CPB have demonstrated that the serum levels of proinflammatory
cytokines, including IL-1, IL-2, IL-4, IL-6, IL-8 and TNF-α are
significantly increased during and following CPB (7–13).
Wan et al (13) summarized
the cytokine responses during clinical CPB and illustrated the
complexity and diversity of the CPB-induced cytokine response. The
present review aimed to focus on the proinflammatory cytokine
TNF-α.
Studies on the effects of CPB on the serum level of
TNF-α in patients are controversial. Zhang et al (10) revealed that the serum level of
TNF-α in patients undergoing CPB significantly increases when CPB
begins and reaches a maximum following CPB. By contrast, Welters
et al (14) and
Martínez-Comendador et al (15) reported that the plasma level of
TNF-α is not statistically different pre- and post-surgery. The
discrepancy may be due to the different trial design and different
experimental procedure in these studies and also the complex
chemistry of TNF-α in solution. The diverse baseline of TNF-α in
different patients, which is associated with the unique genetic
background of each individual may also contribute to this
discrepancy. Previous studies have demonstrated that circulating
TNF-α level in patients correlates with TNF-α polymorphisms. Boehm
et al (16) analyzed two
polymorphisms from the promoter region of the TNF-α gene, TNF-α-863
C/A and TNF-α-308 G/A. They revealed significant associations of
the TNF-α 863 CC variant with higher TNF-α level preoperatively,
following CPB and 6 h postoperatively, while the TNF-α 863 AA
allele correlated with lower TNF-α level at all time points.
Compared with the TNF-α 863 C/A polymorphism, fewer significant
associations were detected between the TNF-α 308 G/A polymorphism
and TNF-α level. The patients with the TNF-α 308 GG allele have
lower TNF-α level immediately following CPB, whereas TNF-α 308 AA
carriers are significantly associated with elevated TNF-α level
preoperatively and immediately following CPB (16). Studies on the association between
TNF-α 308 G/A polymorphism and serum level of TNF-α by other groups
demonstrate varying results. Yoon et al (17) demonstrated that the TNF-α 308 AA
variant correlated with increased TNF-α level, while Galiñanes
et al (18) did not detect
any correlation.
In animal models of CPB in which the genetic
background of the experimental animal is more uniform than in
humans, including rabbit, piglet and rat models, it is consistently
observed that the serum level of TNF-α is significantly increased
during and following CPB (11,12,19,20).
Notably, Qi et al (11)
demonstrated that in a rabbit model of CPB, in addition to a
significant increase of systemic levels of TNF-α, the serum level
of TNF-α from the left atrium is significantly higher than that
from the right atrium (Table I).
Furthermore, TNF-α mRNA level in lung tissue is also increased
markedly during CPB, suggesting that CPB can induce pulmonary TNF-α
synthesis and release (11), which
could further exasperate CPB-induced lung injury.
 | Table ITNF-α level in blood samples
(pg/l). |
Table I
TNF-α level in blood samples
(pg/l).
| Prior to CPB | | 5 min after aortic
clamp release | | CPB
termination | |
|---|
|
| |
| |
| |
|---|
| Group | Right atrium | Left atrium | P-value | Right atrium | Left atrium | P-value | Right atrium | Left atrium | P-value |
|---|
| I | 104.40±14.17 | 111.55±16.98 | P>0.05 | 116.27±32.65 | 116.76±20.33 | P>0.05 | 115.88±30.81 | 110.90±28.43 | P>0.05 |
| II | 110.14±48.32 | 94.45±37.18 | P>0.05 |
240.37±56.44a | 329.24±69.99 | P<0.05 |
208.25±39.15a | 257.30±38.22 | P<0.05 |
| III | 110.61±38.22 | 108.25±22.90 | P>0.05 |
236.73±41.92a | 319.74±48.11 | P<0.05 |
207.35±52.11a | 266.65±40.23 | P<0.05 |
| IV | 110.49± 47.56 | 113.63±33.05 | P>0.05 |
212.98±55.30a | 215.49±56.28 | P>0.05 |
189.84±37.09a | 192.79±39.43 | P>0.05 |
TNF-α mediates lung injury during
CPB
Although direct clinical evidence demonstrating that
TNF-α can cause lung damage during and following CPB remains
lacking, the association between increased level of TNF-α and
pulmonary dysfunction has been indicated in multiple studies.
Dauber et al revealed that peak circulating TNF levels
correlate with CPB-induced coronary and pulmonary vascular injury
(21). Studies on a rabbit model
of CPB demonstrated that endotracheal administration or pulmonary
perfusion of a neutralizing antibody against TNF-α significantly
reduced pulmonary edema and alleviated histological damage in the
lung (11,12). In a rat model, TNF-α markedly
induced pulmonary vascular barrier dysfunction with increased lung
water content and impaired oxygenation (22).
The molecular and cellular mechanisms underlying
TNF-α-mediated lung injury remain to be elucidated. As a
proinflammatory cytokine, TNF-α has been revealed to not only
directly induce apoptosis in pulmonary endothelial and alveolar
epithelial cells but also trigger a cascade of immune reactions to
damage lung function indirectly. Petrache et al used bovine
pulmonary artery endothelial cells to investigate the molecular
mechanism underlying TNF-α-induced endothelial cell apoptosis and
barrier dysfunction (23). They
demonstrated that TNF-α significantly enhanced apoptosis and
stimulated the formation of stress fibers and paracellular gaps by
increasing myosin light chain (MLC) phosphorylation through MLC
kinase and Rho kinase, which consequently resulted in the reduction
of transcellular electrical resistance (23). Wang et al (24) demonstrated that human TNF-α induces
dose-dependent apoptosis in pulmonary alveolar epithelial cells
derived from the human lung carcinoma cell line A549 and primary
cultures of well-differentiated type II alveolar epithelial cells
from the rat. TNF-α-induced apoptosis requires the induction of
angiotensinogen expression, proteolytic processing of the
synthesized angiotensinogen protein and the subsequent binding of
angiotensin II to its receptor.
At the cellular level, TNF-α has been found to
activate and recruit PMNs (25,26).
The activated PMNs secrete proteolytic enzymes, including MMPs and
elastase and release oxygen-free radicals into the systemic
circulation and lung tissue (25).
Consequently, this leads to the degradation of pulmonary
ultrastructure and permeabilization of pulmonary alveolar cells and
endothelium. It has also been demonstrated that TNF-α can regulate
the immune capacity of monocytes and macrophages by regulating the
expression of toll-like receptors (TLRs). Tsai et al
(26) used human monocytic THP-1
cells to elucidate that TNF-α can downregulate TLR4 expression and
induce intracellular tristetraprolin (TTP) expression through the
mitogen-activated protein kinase (MAPK)/extracellular-signal
regulated kinase signaling pathway (Fig. 1). Their results from in
vitro tissue culture are consistent with the observation of
clinical samples, which demonstrate that TTP expression increases
and TLR4 expression decreases in monocytes following CPB (26).
3. Therapeutic strategies targeting TNF-α to
attenuate CPB-induced lung injury
Inhibition of TNF-α production
It has been revealed that p38 MAPK is involved in
regulating TNF-α transcription (27). Thus, inhibition of p38 MAPK may be
an effective approach to reduce TNF-α production. Dong et al
pretreated rats using intravenous administration of the specific
p38 MAPK inhibitor, SB203580 in 0.5 ml saline 30 min prior to
establishing CPB and demonstrated that the pre-treatment not only
significantly reduced the mRNA and protein level of TNF-α in lung
tissue compared with those in rats undergoing CPB without
administration of the inhibitor, but also attenuated lung tissue
water and CPB-mediated damage (28). However, the expression of other
proinflammatory cytokines, including IL-1β is also significantly
reduced by the inhibitor. In addition, the p38 MAPK inhibitor
affects the activity of nuclear factor (NF)-κB, which is also a
TNF-α transcription factor but with less specificity (28). Thus, it appears that inhibition of
TNF-α transcription factors may induce a global effect on
inflammatory reactions during CPB. The possible clinical
application of such inhibitors remains inconclusive.
Inhibition of TNF-α activity
Functional neutralizing antibodies against TNF-α
appear to be a more specific strategy for inhibiting TNF-α activity
compared with the inhibition of TNF-α transcription. Qi et
al (11) and Yu et al
(12) demonstrated that in a
rabbit model of CPB, neutralizing antibody against TNF-α
administered either by endotracheal intubation or by pulmonary
artery perfusion alleviates CPB-induced pulmonary tissue damage,
prevents CPB-induced lung edema, significantly improves oxygenation
index and reduces CPB-induced pulmonary inflammation. In addition
to reducing the serum level of TNF-α, Yu et al demonstrated
that pulmonary artery perfusion with an antibody against TNF-α
markedly decreased TNF-α expression in lung tissue (Table I and Fig. 2) (11,12).
Studies on ovarian cancer cells and keratinocytes revealed that
TNF-α stimulates its own mRNA synthesis in an autocrine manner
(29,30). Thus, functional inhibition of TNF-α
was able to interrupt the autocrine TNF-α synthesis loop and
ultimately reduce TNF-α production. Similar to the study by Yu
et al, Szlosarek et al also demonstrated that the
TNF-α neutralizing antibody infliximab reduces TNF-α mRNA level in
ovarian cancer cells by inhibiting the autocrine production loop
(29). Thus, functional inhibition
of TNF-α appears to be a strategy superior to inhibition of TNF-α
transcription factors since it not only specifically inhibits TNF-α
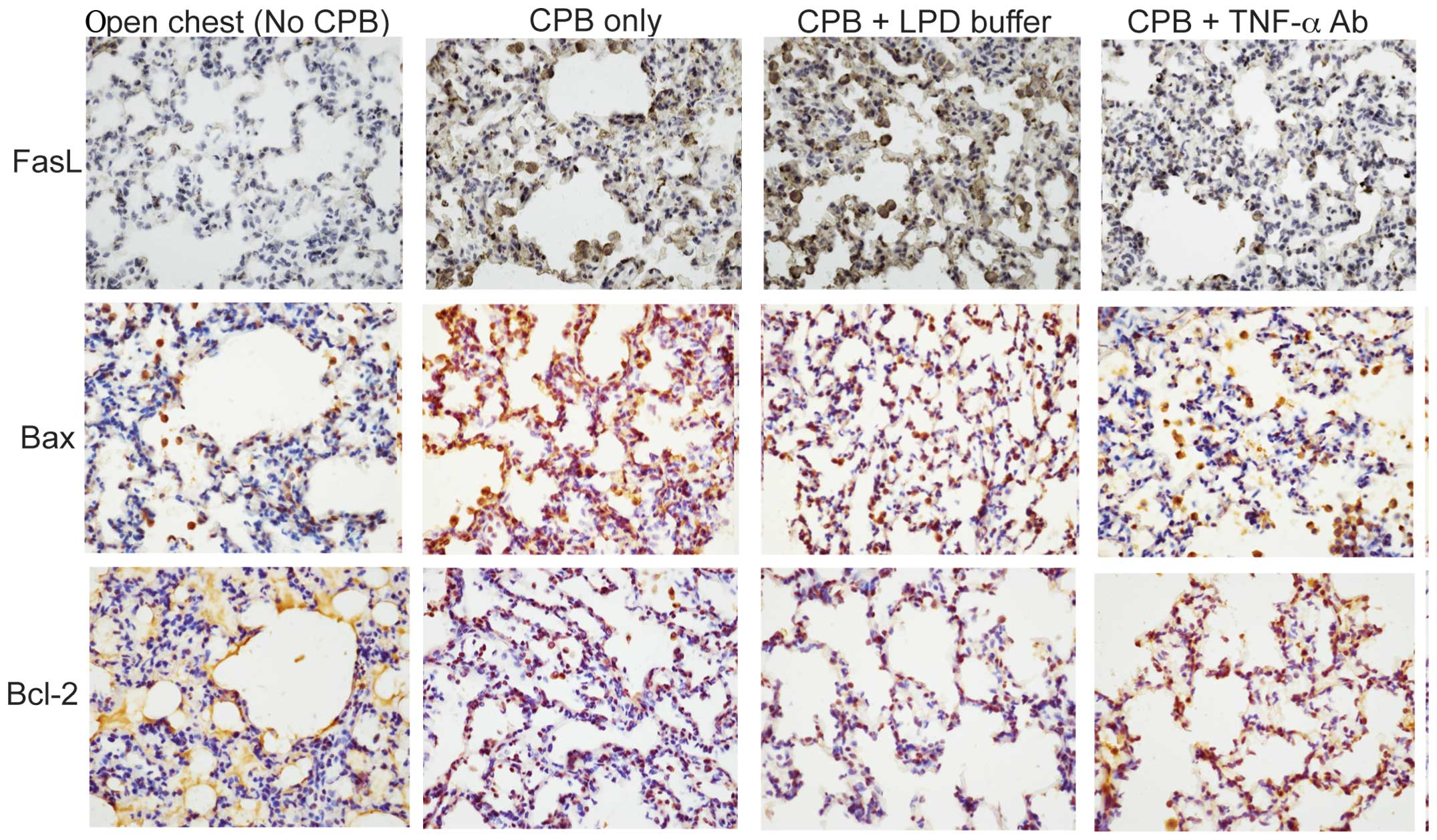
activity but also reduces its production. Yu et al (12) further investigated the molecular
mechanisms underlying TNF-α antibody-mediated attenuation of
PBC-induced lung injury. They revealed that TNF-α antibody
administered either by endotracheal intubation or by pulmonary
artery perfusion significantly reduces CPB-induced pulmonary
apoptosis in a rabbit model of CPB (Fig. 3). In addition, Imai et al
demonstrated that intratracheal anti-TNF-α antibody attenuates
ventilator-induced lung injury in rabbits (31).
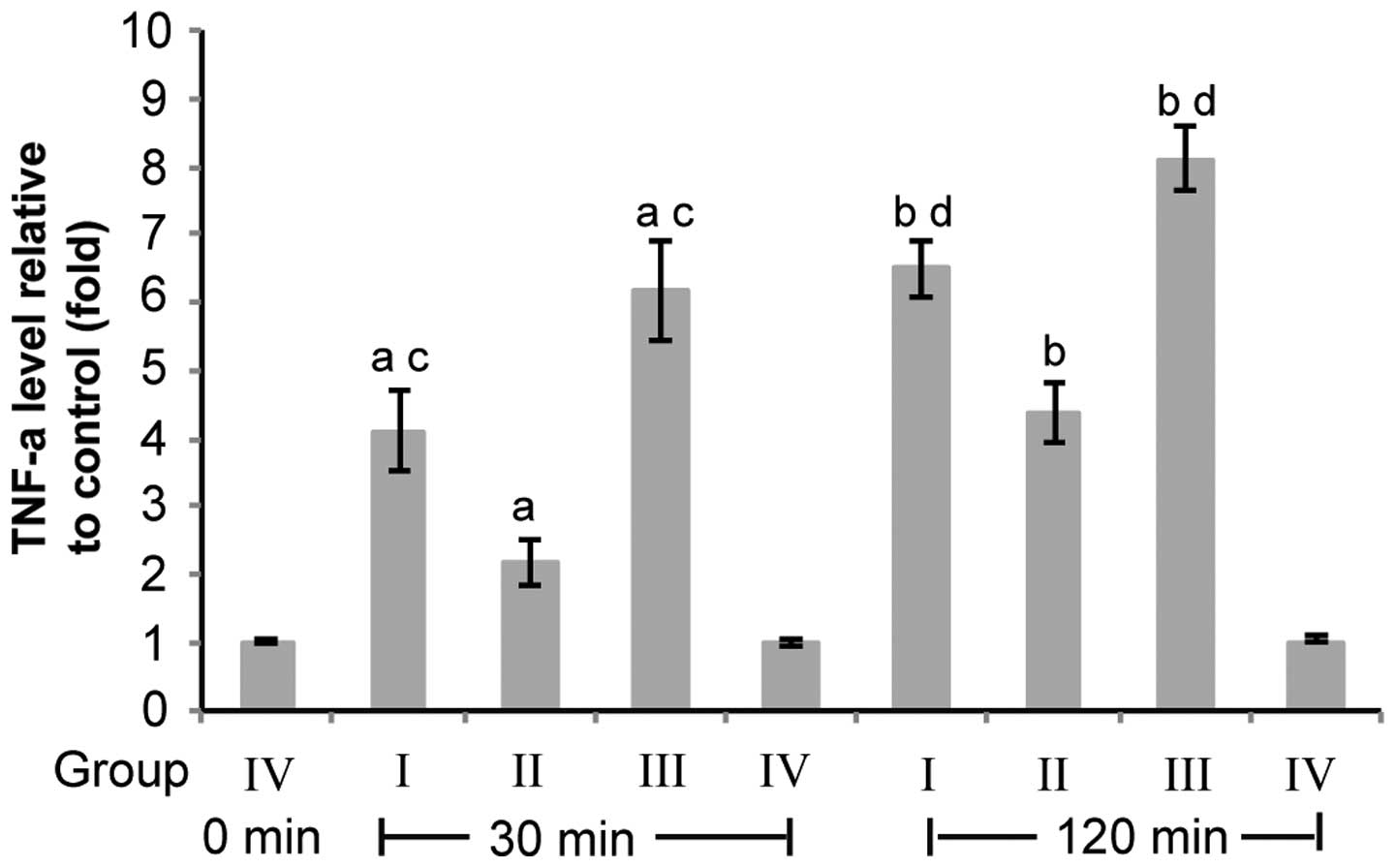 | Figure 2Pulmonary artery perfusion with TNF-α
Ab reduces CPB-induced TNF-α overexpression in lung tissue. Group
I, CPB+perfusion buffer; Group II, CPB+TNF-α Ab; Group III, CPB
only; Group IV, Open heart no CPB. Rabbit lung tissues were
collected following CPB for 30 and 120 min for group I–III or
following the chest being opened for 30 and 120 min for group IV.
β-actin was used as the reference gene. TNF-α level was normalized
to the reference gene in each sample. The TNF-α level relative to
that of the rabbits in group IV was calculated using the equation
2ΔΔCT (n=10). Data are presented as the mean ± standard
deviation. aP<0.05, group I, II and III vs. group IV
for 30 min; bP<0.05, group I, II and III vs. group IV
for 120 min; cP<0.05, group I and III vs. group II
for 30 min; dP<0.05, group I and III vs. group II for
120 min (12). TNF-α, tumor
necrosis factor-α; CPB, cardiopulmonary bypass; TLR, toll-like
receptor; Ab, antibody. |
TNF-α has been demonstrated to bind at least two
receptors, TNFRI-p55 and TNFRII-p75 (32). The extracellular domain of these
receptors can be shed into the circulation as soluble TNF-α
receptors p55 and p75. Elevation of TNF-α level increases the
levels of soluble receptors, which are generated by the cleavage of
the membrane-bound TNF receptors by a metalloproteinase after TNF
binds to its membrane-associated receptors (33). Elevated soluble TNF-α receptor
concentrations have been revealed to be an indication of a systemic
inflammatory response (34). The
binding of the soluble receptors to TNF-α inhibits the biological
activity and prolongs the half-life of circulating TNF-α (35). The inhibitory effect of soluble
receptors on TNF-α activity has been demonstrated in an
investigation into rheumatoid arthritis. Wooley et al
demonstrated that a recombinant human TNF receptor, Fc-fusion
protein significantly reduced the incidence and the severity of
collagen-induced arthritis in mice (36). Thus, application of soluble
receptors to inhibit TNF-α activity appears to be another rational
strategy for attenuating TNF-α-mediated lung injury during CPB.
The clinical application of antibodies against TNF-α
and the soluble TNF-α receptors p55 and p75 has been extensively
investigated in human diseases where TNF-α has a critical role in
disease development and progression, including sepsis and
rheumatoid arthritis. They have been assessed in clinical trials of
sepsis treatment. In phase I-III clinical trials, such agents
appear to be safe and non-antigenic when they are administered
intravenously to septic patients. However, the results of the
sepsis trials varied significantly. Thus, the efficacy of TNF-α
antibody and the soluble TNF-α receptors to treat human sepsis
remains inconclusive. By contrast, the application of those agents
in human rheumatoid arthritis has been demonstrated to be
effective. In treating arthritis, these agents appear safe, improve
disease symptoms in a dose-dependent manner and markedly reduce
erythrocyte sedimentation rate and C-reactive protein level, which
are indicators of systemic inflammation (37). Therefore, based on the results from
studies on the rabbit model of CPB demonstrating that anti-TNF-α
antibody can significantly attenuate CPB-induced lung injury
(11,12) and the successful clinical
application of TNF-α antibodies and soluble TNF-α receptors in
human rheumatoid arthritis, it appears that anti-TNF-α antibodies
and soluble TNF-α receptors could be promising therapeutic
strategies for relief of the pulmonary dysfunction associated with
CPB.
4. Effects of anti-inflammatory intervention
on TNF-α level
In addition to the strategies directly targeting
TNF-α, other anti-inflammatory interventions aimed at alleviating
CPB-induced lung injury have been revealed to reduce TNF-α level.
For instance, modification of artificial circuits, including
heparin-coated and phosphorylcholine-coated circuits are found to
significantly reduce systemic TNF-α level (38,39).
Pharmacological interventions also appear to effectively inhibit
TNF-α production during and following CPB. Administration of
corticosteroids prior to CPB or aprotinin following CPB reduces the
production of TNF-α and other proinflammatory cytokines (40), while the antibiotic agents
moxifloxacin and cefuroxime, which have been demonstrated to
inhibit cytokine release from monocytes and neutrophils, do not
attenuate the inflammatory cytokine response to CPB (41).
Statin drugs, which are increasingly recognized as
having anti-inflammatory effects, have been widely investigated for
their potential protective effects on patients undergoing CPB.
Morgan et al published a systematic review of clinical
trials examining the clinical benefits of pre-operative
prophylactic statin therapy for patients undergoing CPB (42). Although the pooled data in their
review support that pre-operative treatment with statins attenuates
CPB-induced release of proinflammatory cytokines, including IL-6,
IL-8 and TNF- α in patients, due to the limited number of studies
and lack of rigorous study design in certain trials, the potential
anti-inflammatory effects of pre-operative statin therapy remain
inconclusive.
5. Conclusion
In the present review, the effects of CPB on TNF-α
level in the serum and lung tissue in patients and animal models of
CPB were summarized, the molecular and cellular mechanisms
underlying TNF-α-mediated lung injury during CPB were discussed and
several therapeutic strategies targeting TNF-α to attenuate
CPB-induced pulmonary dysfunction were proposed. Although it
remains to be elucidated how serum TNF-α level correlates with
CPB-associated morbidity in patients and whether serum TNF-α level
can predict post-CPB complications, it appears that pulmonary
artery perfusion with neutralizing antibody for TNF-α, which can
specifically inhibit pulmonary TNF-α production and activity, may
be a superior strategy to attenuate CPB-induced lung injury.
The functions of other cytokines, including IL-6,
IL-8 and IL-10 must not be ignored. The production and activity of
these cytokines are also significantly affected by CPB. TNF-α and
other cytokines may act synergistically, ultimately resulting in
pulmonary dysfunction during CPB. Thus, restoration of the
homeostasis of proinflammatory and anti-inflammatory cytokines may
be the key to reducing CPB-induced lung injury.
References
|
1
|
Stamou SC, Pfister AJ, Dangas G, et al:
Beating heart versus conventional single-vessel reoperative
coronary artery bypass. Ann Thorac Surg. 69:1383–1387. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Asimakopoulos G, Smith PL, Ratnatunga CP
and Taylor KM: Lung injury and acute respiratory distress syndrome
after cardiopulmonary bypass. Ann Thorac Surg. 68:1107–1115. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hammermeister KE, Burchfiel C, Johnson R
and Grover FL: Identification of patients at greatest risk for
developing major complications at cardiac surgery. Circulation.
82:380–389. 1990.
|
|
4
|
Ng CS, Wan S, Yim AP and Arifi AA:
Pulmonary dysfunction after cardiac surgery. Chest. 121:1269–1277.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Taggart DP, el-Fiky M, Carter R, Bowman A
and Wheatley DJ: Respiratory dysfunction after uncomplicated
cardiopulmonary bypass. Ann Thorac Surg. 56:1123–1128. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wan S, LeClerc JL and Vincent JL:
Inflammatory response to cardiopulmonary bypass: mechanisms
involved and possible therapeutic strategies. Chest. 112:676–692.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diegeler A, Doll N, Rauch T, Haberer D,
Walther T, Falk V, Gummert J, Autschbach R and Mohr FW: Humoral
immune response during coronary artery bypass grafting: a
comparison of limited approach, ‘off-pump’ technique, and
conventional cardiopulmonary bypass. Circulation. 102(Suppl):
III95–III100. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ascione R, Lloyd CT, Underwood MJ, Lotto
AA, Pitsis AA and Angelini GD: Inflammatory response after coronary
revascularization with or without cardiopulmonary bypass. Ann
Thorac Surg. 69:1198–1204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matata BM, Sosnowski AW and Galiñanes M:
Off-pump bypass graft operation significantly reduces oxidative
stress and inflammation. Ann Thorac Surg. 69:785–791. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Z, Wu Y, Zhao Y, Xiao X, Liu J and
Zhou X: Dynamic changes in HMGB1 levels correlate with inflammatory
responses during cardiopulmonary bypass. Exp Ther Med. 5:1523–1527.
2013.PubMed/NCBI
|
|
11
|
Qi D, Gao MX and Yu Y: Intratracheal
antitumor necrosis factor-α antibody attenuates lung tissue damage
following cardiopulmonary bypass. Artif Organs. 37:142–149. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu Y, Gao M, Li H, Zhang F and Gu C:
Pulmonary artery perfusion with anti-tumor necrosis factor alpha
antibody reduces cardiopulmonary bypass-induced inflammatory lung
injury in a rabbit model. PLoS One. 8:e832362013. View Article : Google Scholar
|
|
13
|
Wan S, LeClerc JL and Vincent JL: Cytokine
responses to cardiopulmonary bypass: lessons learned from cardiac
transplantation. Ann Thorac Surg. 63:269–276. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Welters ID, Feurer MK, Preiss V, Müller M,
Scholz S, Kwapisz M, Mogk M and Neuhäuser C: Continuous
S-(+)-ketamine administration during elective coronary artery
bypass graft surgery attenuates pro-inflammatory cytokine response
during and after cardiopulmonary bypass. Br J Anaesth. 106:172–179.
2011. View Article : Google Scholar
|
|
15
|
Martínez-Comendador JM, Alvarez JR,
Mosquera I, Sierra J, Adrio B, Carro JG, Fernández A and Bengochea
J: Preoperative statin treatment reduces systemic inflammatory
response and myocardial damage in cardiac surgery. Eur J
Cardiothorac Surg. 36:998–1005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boehm J, Hauner K, Grammer J, Dietrich W,
Wagenpfeil S, Braun S, Lange R and Bauernschmitt R: Tumor necrosis
factor-α-863 C/A promoter polymorphism affects the inflammatory
response after cardiac surgery. Eur J Cardiothorac Surg.
40:e50–e54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yoon SZ, Jang IJ, Choi YJ, Kang MH, Lim
HJ, Lim YJ, Lee HW, Chang SH and Yoon SM: Association between tumor
necrosis factor alpha 308G/A polymorphism and increased
proinflammatory cytokine release after cardiac surgery with
cardiopulmonary bypass in the Korean population. J Cardiothorac
Vasc Anesth. 23:646–650. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Galiñanes M, James M, Codd V, Baxi A and
Hadjinikolaou L: TNF-alpha gene promoter polymorphism at nucleotide
-308 and the inflammatory response and oxidative stress induced by
cardiac surgery: role of heart failure and medical treatment. Eur J
Cardiothorac Surg. 34:332–337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yewei X, Liya D, Jinghao Z, Rufang Z and
Li S: Study of the mechanism of pulmonary protection strategy on
pulmonary injury with deep hypothermia low flow. Eur Rev Med
Pharmacol Sci. 17:879–885. 2013.PubMed/NCBI
|
|
20
|
Shen Y, Wu H, Wang C, Shao H, Huang H,
Jing H and Li D: Simvastatin attenuates cardiopulmonary
bypass-induced myocardial inflammatory injury in rats by activating
peroxisome proliferator-activated receptor γ. Eur J Pharmacol.
649:255–262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dauber IM, Parsons PE, Welsh CH, Giclas
PC, Whitman GJ, Wheeler GS, Horwitz LD and Weil JV: Peripheral
bypass-induced pulmonary and coronary vascular injury. Association
with increased levels of tumor necrosis factor. Circulation.
88:726–735. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Worrall NK, Chang K, LeJeune WS, Misko TP,
Sullivan PM, Ferguson TB Jr and Williamson JR: TNF-alpha causes
reversible in vivo systemic vascular barrier dysfunction via
NO-dependent and -independent mechanisms. Am J Physiol.
273:H2565–H2574. 1997.
|
|
23
|
Petrache I, Verin AD, Crow MT, Birukova A,
Liu F and Garcia JG: Differential effect of MLC kinase in
TNF-alpha-induced endothelial cell apoptosis and barrier
dysfunction. Am J Physiol Lung Cell Mol Physiol. 280:L1168–L1178.
2001.PubMed/NCBI
|
|
24
|
Wang R, Alam G, Zagariya A, Gidea C,
Pinillos H, Lalude O, Choudhary G, Oezatalay D and Uhal BD:
Apoptosis of lung epithelial cells in response to TNF-alpha
requires angiotensin II generation de novo. J Cell Physiol.
185:253–259. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Faymonville ME, Pincemail J, Duchateau J,
Paulus JM, Adam A, Deby-Dupont G, Deby C, Albert A, Larbuisson R,
Limet R, et al: Myeloperoxidase and elastase as markers of
leukocyte activation during cardiopulmonary bypass in humans. J
Thorac Cardiovasc Surg. 102:309–317. 1991.PubMed/NCBI
|
|
26
|
Tsai CS, Chen DL, Lin SJ, Tsai JC, Lin TC,
Lin CY, Chen YH, Huang GS, Tsai HY, Lin FY and Li CY: TNF-alpha
inhibits toll-like receptor 4 expression on monocytic cells via
tristetraprolin during cardiopulmonary bypass. Shock. 32:40–48.
2009. View Article : Google Scholar
|
|
27
|
Lee JC and Young PR: Role of CSB/p38/RK
stress response kinase in LPS and cytokine signaling mechanisms. J
Leukoc Biol. 59:152–157. 1996.PubMed/NCBI
|
|
28
|
Dong X, Liu Y, Du M, Wang Q, Yu CT and Fan
X: P38 mitogen-activated protein kinase inhibition attenuates
pulmonary inflammatory response in a rat cardiopulmonary bypass
model. Eur J Cardiothorac Surg. 30:77–84. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Szlosarek PW, Grimshaw MJ, Kulbe H, Wilson
JL, Wilbanks GD, Burke F and Balkwill FR: Expression and regulation
of tumor necrosis factor alpha in normal and malignant ovarian
epithelium. Mol Cancer Ther. 5:382–390. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lisby S, Faurschou A and Gniadecki R: The
autocrine TNFalpha signalling loop in keratinocytes requires
atypical PKC species and NF-kappaB activation but is independent of
cholesterol-enriched membrane microdomains. Biochem Pharmacol.
73:526–533. 2007. View Article : Google Scholar
|
|
31
|
Imai Y, Kawano T, Iwamoto S, Nakagawa S,
Takata M and Miyasaka K: Intratracheal anti-tumor necrosis
factor-alpha antibody attenuates ventilator-induced lung injury in
rabbits. J Appl Physiol (1985). 87:510–515. 1999.
|
|
32
|
Torre-Amione G, Kapadia S, Lee J, Bies RD,
Lebovitz R and Mann DL: Expression and functional significance of
tumor necrosis factor receptors in human myocardium. Circulation.
92:1487–1494. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Katsura K, Park M, Gatanaga M, Yu EC,
Takishima K, Granger GA and Gatanaga T: Identification of
proteolytic enzyme which cleaves human p75 TNF receptor in vitro.
Biochem Biophys Res Commun. 222:298–302. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lantz M, Malik S, Slevin ML and Olsson I:
Infusion of tumor necrosis factor (TNF) causes an increase in
circulating TNF-binding protein in humans. Cytokine. 2:402–406.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kapadia S, Torre-Amione G, Yokoyama T and
Mann DL: Soluble TNF binding proteins modulate the negative
inotropic properties of TNF-alpha in vitro. Am J Physiol.
268:H517–H525. 1995.PubMed/NCBI
|
|
36
|
Wooley PH, Dutcher J, Widmer MB and Gillis
S: Influence of a recombinant human soluble tumor necrosis factor
receptor FC fusion protein on type II collagen-induced arthritis in
mice. J Immunol. 151:6602–6607. 1993.PubMed/NCBI
|
|
37
|
Cain BS, Meldrum DR, Harken AH and
McIntyre RC Jr: The physiologic basis for anticytokine clinical
trials in the treatment of sepsis. J Am Coll Surg. 186:337–350.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamada H, Kudoh I, Hirose Y, Toyoshima M,
Abe H and Kurahashi K: Heparin-coated circuits reduce the formation
of TNF alpha during cardiopulmonary bypass. Acta Anaesthesiol
Scand. 40:311–317. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schulze CJ, Han L, Ghorpade N, Etches WS,
Stang L, Koshal A and Wang SH: Phosphorylcholine-coated circuits
improve preservation of platelet count and reduce expression of
proinflammatory cytokines in CABG: a prospective randomized trial.
J Card Surg. 24:363–368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Apostolakis EE, Koletsis EN, Baikoussis
NG, Siminelakis SN and Papadopoulos GS: Strategies to prevent
intraoperative lung injury during cardiopulmonary bypass. J
Cardiothorac Surg. 5:12010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wiesner G, Braun SL, Gruber M, Gertler R,
Lange R, Tassani P and Martin K: Neither moxifloxacin nor
cefuroxime produces significant attenuation of inflammatory
mediator release in patients exposed to cardiopulmonary bypass: a
randomized controlled trial. J Antimicrob Chemother. 67:230–233.
2012. View Article : Google Scholar
|
|
42
|
Morgan C, Zappitelli M and Gill P: Statin
prophylaxis and inflammatory mediators following cardiopulmonary
bypass: a systematic review. Crit Care. 13:R1652009. View Article : Google Scholar : PubMed/NCBI
|