Introduction
Transient global cerebral ischemia, due to temporary
blood flow deprivation of the brain, causes insidious delayed
neuronal degeneration in the hippocampus (1,2).
Specifically, the pyramidal neurons in the hippocampal Cornu
Ammonis region CA1 are the most vulnerable to transient global
cerebral ischemia; however, neurons in the CA3 and dentate gyrus
remain essentially intact (1,3).
Neuronal death in the CA1 region, which occurs several days after
ischemia-reperfusion (I-R), is described as ‘delayed neuronal
death’ (1). It has been suggested
that the molecular events associated with delayed neuronal death
are caused by glutamate receptor-mediated neurotoxicity (4), free radical-related damage (5) and oxidative stress (6). However, the precise mechanisms of
delayed neuronal death remain unclear.
Inhibitors of DNA binding/differentiation (ID)
proteins regulate gene transcription through binding to basic
helix-loop-helix (bHLH) transcription factors; four members of this
protein family, ID1–4, have been identified in mammals (7–10).
Members of the ID protein family share a highly conserved bHLH
domain and are similar in size (13–20 kDa), but display extensive
sequence variation outside the bHLH domain. As transcription
factors, ID proteins are involved in the development of the nervous
system, muscle genesis, tumorigenesis, cell cycle regulation and
apoptosis (10–12). ID1–3 are expressed in dividing
neuroblasts of the central nervous system (CNS) during development
(13,14). However, ID4, which is dissimilar to
the other ID proteins, is exclusively localized in the regions
undergoing neuronal maturation in the CNS and the peripheral
nervous system (13,15). Expression of ID proteins is very
limited in the adult CNS, but is detectable in distinct populations
of adult post-mitotic neurons in specific regions, such as all
layers of the cerebral cortex except layer IV, the Purkinje cell
layer of the cerebellum, the olfactory bulb (the mitral cell,
glomerular and internal granule cell layers), the hippocampus and
the suprachiasmatic nucleus in the adult rodent brain (14,16–19).
The roles and the changes in ID protein levels
induced by transient cerebral ischemia in the hippocampus have not
been studied in detail. Therefore in the present study, we examined
the changes in immunoreactivity and protein levels of ID1–4 in the
ischemic hippocampus of the gerbil 5 min after transient ischemia;
the gerbil has been established as a good model for the study of
transient global cerebral ischemia (20–23).
Materials and methods
Animals and ethics
Male Mongolian gerbils (Meriones
unguiculatus) were obtained from the Experimental Animal Center
at the Kangwon National University (Chuncheon, Korea). Gerbils were
used at 6 months of age (body weight, 65–75 g). The animals were
housed in conventional cages at 23°C and 60% humidity, with a 12-h
light/12-h dark cycle. The animals had free access to food and
water. The procedures for animal handling and care adhered to
guidelines that are in compliance with the current international
laws and policies (Guide for the Care and Use of Laboratory
Animals, The National Academies Press, 8th edition, Washington DC,
USA, 2011) and they were approved by the Institutional Animal Care
and Use Committee of the Kangwon National University. All
experiments were conducted with care to minimize the number and the
suffering of animals.
Induction of transient cerebral
ischemia
Cerebral ischemia was established with a method
previously described by our group (24,25).
Briefly, ischemia was induced by bilateral common carotid artery
occlusion under anesthesia by inhalation of 2.5% isoflurane in 30%
O2 and 70% N2. Bilateral common carotid
arteries were occluded using non-traumatic aneurysm clips for 5
min. The complete interruption of blood flow was confirmed by
observing the central artery in the retinae using an
ophthalmoscope. During surgery, the animals were kept on a heating
pad at 37±0.5°C. Thereafter, the animals were kept on a thermal
incubator (Mirae Medical Foundation Health Improvement Centre,
Seoul, Korea) to maintain their body temperature until sacrifice.
Sham-operated animals were exposed to similar surgery without
carotid artery occlusion.
Tissue processing for histology
For histological examination, the sham-operated and
ischemic gerbils (sham and ischemia groups, n=7 in each) were
deeply anesthetized with chloral hydrate (300 mg/kg,
intraperitoneal injection) and transcardially perfused with 0.1 M
phosphate-buffered saline (PBS; pH 7.4) followed by perfusion with
4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at 12 h, 1,
2, 5 and 10 days after ischemia-reperfusion. Following perfusion,
the brains were carefully dissected and post-fixed in 4%
paraformaldehyde for 6 h. The brain tissues were cryoprotected by
infiltration in 30% sucrose overnight. Next, frozen tissues
containing the hippocampus were serially cut into 30-μm-thick
slices with a cryostat (CM1900 UV; Leica, Wetzlar, Germany) and
placed into six-well plates containing PBS.
Assessment of neuronal damage
Neuronal damage in the hippocampal CA1 region was
examined in the sham and ischemia groups after transient cerebral
ischemia by Cresyl violet (CV) staining, neuronal nuclear antigen
(NeuN) immunohistochemistry and Fluoro-Jade B (F-J B)
histofluorescence at designated time points (1, 2, 5 and 10 days
after reperfusion).
CV staining
To examine neuronal damage in the brain upon
transient cerebral ischemia, sections from the sham and the
ischemia groups were mounted on gelatin-coated microscopy slides.
Cresyl violet acetate (Sigma-Aldrich, St. Louis, MO, USA) was
dissolved in distilled water at 1.0% (w/v), and glacial acetic acid
was added to this solution. The sections were stained and
dehydrated by immersing in serial ethanol baths, and were then
mounted with Canada balsam (Kanto Chemical Co., Ltd., Tokyo,
Japan).
NeuN immunohistochemical detection
To investigate the neuronal changes in the CA1
region upon transient cerebral ischemia, the anti-NeuN antibody was
used, which targets a neuron-specific soluble nuclear antigen.
Briefly, the sections were sequentially treated with 0.3% hydrogen
peroxide (H2O2) in PBS for 30 min and 10%
normal goat serum in 0.05 M PBS for 30 min. The sections were next
incubated with diluted mouse anti-NeuN (1:1,000; Chemicon
International, Temecula, CA, USA) overnight at 4°C. Next, the
tissues were exposed to biotinylated goat anti-mouse IgG and
streptavidin peroxidase complex (1:200; Vector Laboratories Inc.,
Burlingame, CA, USA). They were then visualized by addition of
3,3′-diaminobenzidine in a 0.1 M Tris-HCl buffer, and mounted on
gelatin-coated slides. Following dehydration, the sections were
mounted with Canada balsam (Kanto Chemical Co., Ltd.).
F-J B histofluorescence staining
F-J B histofluorescence staining procedures were
conducted according to the method reported by Candelario-Jalil
et al (6). Briefly, the
sections were first immersed in a solution containing 1% sodium
hydroxide in 80% alcohol, followed by immersion in 70% alcohol.
They were then transferred to a 0.06% potassium permanganate
solution and transferred to a 0.0004% F-J B staining solution
(Histo-Chem Inc., Jefferson, AR, USA). After washing, the sections
were placed on a slide warmer (~50°C) and then examined using an
epifluorescent microscope (Carl Zeiss, Gottingen, Germany) with a
blue (450–490 nm) excitation light and a barrier filter. With this
method, neurons that undergo degeneration brightly fluoresce in
comparison to the background (26).
Cell counts
In order to ensure objectivity, all measurements
were blindly performed by two observers for each experiment, under
the same conditions. The studied tissue sections were selected in a
120-μm interval based on anatomical landmarks corresponding to an
anteroposterior position −1.4 ~ −1.8 mm from the stereotaxic atlas
of the gerbil brain (27), and
cell counts were obtained by averaging the counts from 20 sections
taken from each animal. NeuN- and F-J B-positive (+)
cell structures were observed from 3 layers of the hippocampus
proper (strata oriens, pyramidal and radiatum) using an AxioM1
light microscope (Carl Zeiss) equipped with a digital camera
(Axiocam; Carl Zeiss) connected to a PC monitor. The number of
NeuN- and F-J B+ cells was counted in a 250×250
μm2 area at approximately the center of the CA1 region.
Cell counts were obtained by averaging the total cell number from
each animal per group.
Immunohistochemical detection of ID
proteins
To obtain accurate immunoreactivity data, sections
from the sham-operated and ischemic animals (n=7 at each time
point) were used at designated time points (0, 12 h, 1, 2, 5 and 10
days after reperfusion) under the same conditions.
Immunohistochemical staining was performed using anti-ID1, -ID2,
-ID3 and ID4 primary antibodies (1:200; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA). In order to establish the specificity
of immunostaining, a negative control test was carried out using
pre-blocking with goat serum, instead of the primary antibody. The
negative control showed no immunoreactivity in the studied
samples.
Twenty sections per animal were selected to
quantitatively analyze immunoreactivity for the ID1, ID2, ID3 and
ID4. Digital images of the hippocampal region were captured under
an AxioM1 light microscope equipped with an Axiocam digital camera
connected to a PC monitor. The immunostaining intensities were
semi-quantitatively evaluated using the MetaMorph 4.01 digital
image analysis software (Universal Imaging Corporation Ltd.,
Marlow, UK). The level of immunoreactivity was scaled as −, ±, +,
or ++, representing no staining ( grey scale value: ≥200), weakly
positive (grey scale value: 150–199), moderate (grey scale value:
100–149), or strong (grey scale value: ≤99) staining, respectively,
as in (28).
Double immunofluorescence staining
In order to identify the cell type showing ID1 and
ID4 immunoreactivity, the sections were processed at 5 days after
surgery inducing ischemia by double immunofluorescence staining. We
used rabbit anti-ID1 (1:25; Santa Cruz Biotechnology, Inc.)/goat
anti-glutamic acid decarboxylase 67 (GAD67) (1:50; Chemicon
International) to detect the γ-aminobutyric-acid (GABA)ergic
neurons, rabbit anti-ID4 (1:25; Santa Cruz Biotechnology,
Inc.)/mouse anti-glial fibrillary acidic protein (GFAP) (1:200;
Chemicon International) to detect the astrocytes, and mouse
anti-ionized calcium-binding adapter molecule 1 (Iba-1) (1:200;
Wako Pure Chemical Industries, Ltd., Osaka, Japan) in order to
detect the microglia. The sections were incubated in the antisera
mixture overnight at room temperature. After washing 3 times for 10
min with PBS, the sections were incubated in a mixture of
fluorescein isothiocyanate-conjugated anti-rabbit IgG (1:600;
Jackson ImmunoResearch, West Grove, PA, USA) and Cy3-conjugated
anti-goat or anti-mouse IgG (1:200; Jackson ImmunoResearch) for 2 h
at room temperature. The sections were then observed under a
confocal microscope (LSM510 META NLO; Carl Zeiss).
Detection of ID1 and ID4 by western blot
analysis
To examine changes in the ID1 and ID4 protein levels
in the hippocampal CA1 region upon transient cerebral ischemia,
sham-operated and ischemic animals (n=5 at each time point) were
used for western blot analysis at designated time points (2 and 5
days after I-R). Following animal sacrifice, the brain was removed
and transversely cut into serial sections of 400-μm thickness using
a vibratome (Leica); the hippocampal CA1 region was then dissected
with a surgical blade. The tissues were homogenized in 50 mmol/l
PBS (pH 7.4) containing 0.1 mmol/l ethylene
glycol-O-O′-bis(2-amino-ethyl)-N,N,N′,N′-tetraacetic acid (pH 8.0),
0.2% Nonidet P-40, 10 mmol/l ethylenediamime-N,N,N′,N′-tetraacetic
acid (pH 8.0), 15 mmol/l sodium pyrophosphate, 100 mmol/l
β-glycerophosphate, 50 mmol/l NaF, 150 mmol/l NaCl, 2 mmol/l sodium
orthovanadate, 1 mmol/l phenylmethylsulfonyl fluoride and 1 mmol/l
dithiothreitol (DTT). Following centrifugation at 22,000 × g, the
protein level was determined in the supernatants using a Pierce™
Micro BCA™ Protein Assay kit with bovine serum albumin as the
standard (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Aliquots containing 20 μg of total protein were
boiled in loading buffer containing 150 mmol/l Tris-HCl (pH 6.8), 3
mmol/l DTT, 6% sodium dodecyl sulphate, 0.3% bromophenol blue and
30% glycerol. The aliquots were then loaded onto a 10%
polyacrylamide gel. Following electrophoresis, the gels were
transferred onto nitrocellulose transfer membranes (Pall Corp.,
East Hills, NY, USA). To reduce background staining, the membranes
were incubated with 5% non-fat dry milk in PBS containing 0.1%
Tween-20 for 45 min, followed by incubation with rabbit anti-ID1
and -ID4 antisera (1:1,000), peroxidase-conjugated goat anti-rabbit
IgG (Sigma-Aldrich) and a Pierce™ Enhanced Chemiluminescent (ECL)
substrates (32106; Thermo Fisher Scientific Inc.). The western
blots were scanned, and densitometric analysis for the
quantification of the bands was performed using the Scion Image
software (Scion Corp., Frederick, MD, USA), which provided measures
of relative optical density (ROD). ROD values were expressed as a
percentage; the ROD of the sham group was defined as 100%.
Statistical analysis
Data were expressed as the mean ± standard error of
the mean (SEM). Differences in the mean ROD between groups were
statistically evaluated by a one-way analysis of variance (ANOVA)
using the SPSS program (IBM, Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Neuronal cell death
In this study, we examined delayed neuronal death in
the hippocampal CA1 region using CV histochemistry, NeuN
immunohistochemistry and F-J B histofluorescence staining (Fig. 1). In the sham-operated group,
neurons in the stratum pyramidale of the CA1 region were well
stained with CV and NeuN, but not with F-J B (Table I, Fig.
1A–D). One and two days following I-R, we did not find any
significant change in the number of CV+,
NeuN+ and F-J B+ cells in the ischemic CA1
region (Fig. 1E–H). However, 5
days after I-R, CV+ cells and NeuN+ neurons
were significantly decreased in the stratum pyramidale of the CA1
region (Table I, Fig. 1I–K); at this time point, numerous
F-J B+ cells were observed in the stratum pyramidale of
the CA1 region (Table I, Fig. 1L). At 10 days following I-R, the
distribution pattern of CV+ cells, NeuN+
neurons and F-J B+ cells in the ischemic CA1 region was
similar to that observed at the 5 days post-ischemia (Fig. 1M–P). In the CA2 and CA3 regions, no
difference in the distribution pattern of CV+ neurons
was found between the sham-operated and the ischemia groups
(Fig. 1A, E, I and M).
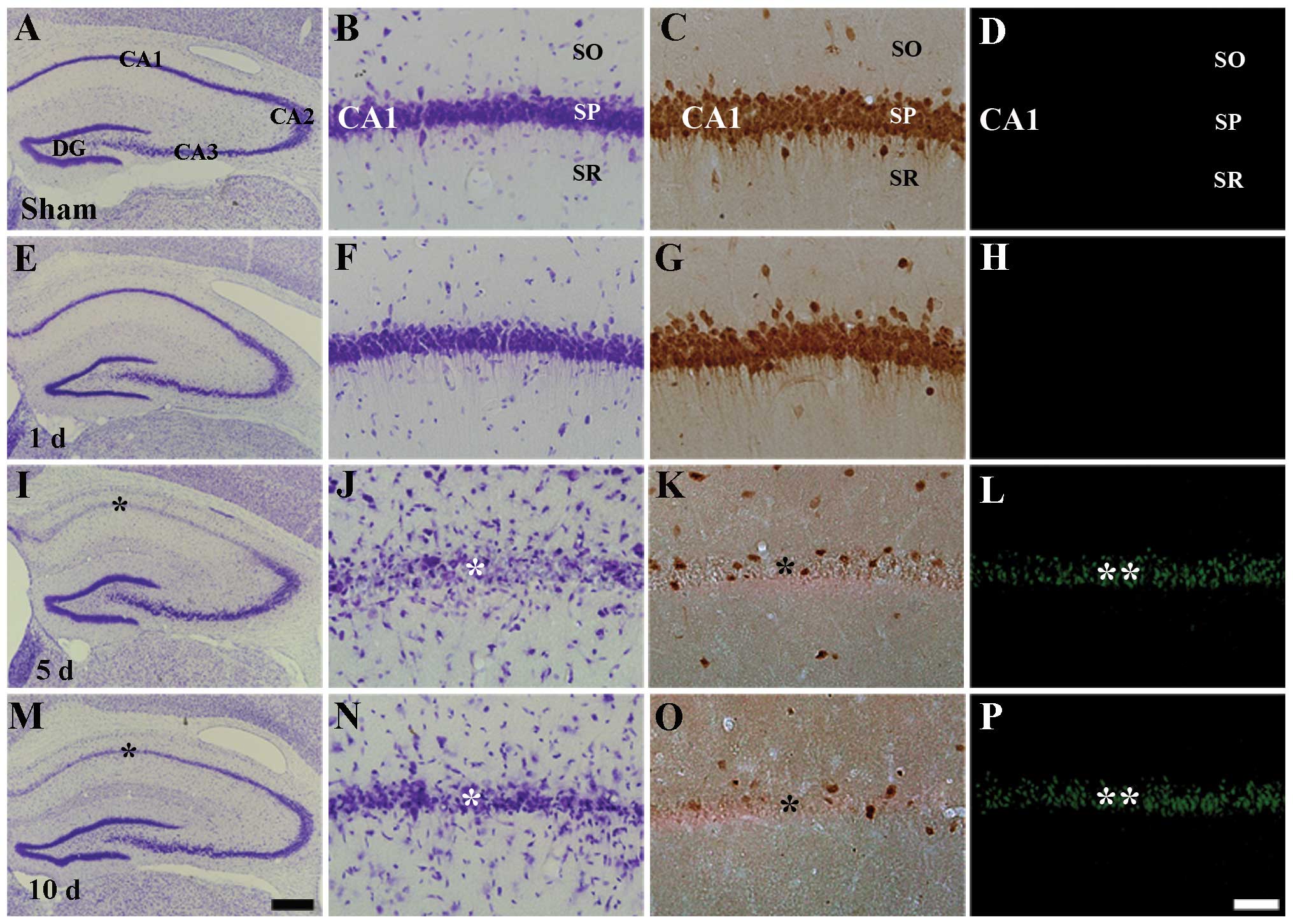 | Figure 1Cresyl violet (CV) staining (first
and second columns), neuronal nuclear antigen (NeuN)
immunohistochemical staining (third column) and Fluoro-Jade B (F-J
B) histofluorescence staining (fourth column) in the Cornu
Ammonis regions CA1 (A-D) of the sham-operated and (E-P)
ischemia groups. In the sham-operated group, numerous
CV+, NeuN+, and no F-J B+ cells
are detected in the stratum pyramidale (SP). In the ischemia
groups, only a few CV+ (black asterisks) and
NeuN+ neurons (arrowhead), and numerous F-J
B+ cells (white asterisks) are detected in the stratum
pyramidale (SP) at 5 and 10 days following ischemia-reperfusion.
DG, dentate gyrus; SO, stratum oriens; and SR, stratum radiatum.
Scale bar=800 (A, E, I and M) and 50 μm (B-D, F-H, J-L and
N-P). |
 | Table IChanges in the mean number of
positively-stained cells in the pyramidal neurons of the ischemic
hippocampal Cornu Ammonis region CA1 of the gerbil. |
Table I
Changes in the mean number of
positively-stained cells in the pyramidal neurons of the ischemic
hippocampal Cornu Ammonis region CA1 of the gerbil.
| Positive cells |
|---|
|
|
|---|
| Time after I-R |
NeuN+ | F-J
B+ |
|---|
| Sham | 364±17.74 | 0 |
| 5 d | 39±10.36a | 207±28.66a |
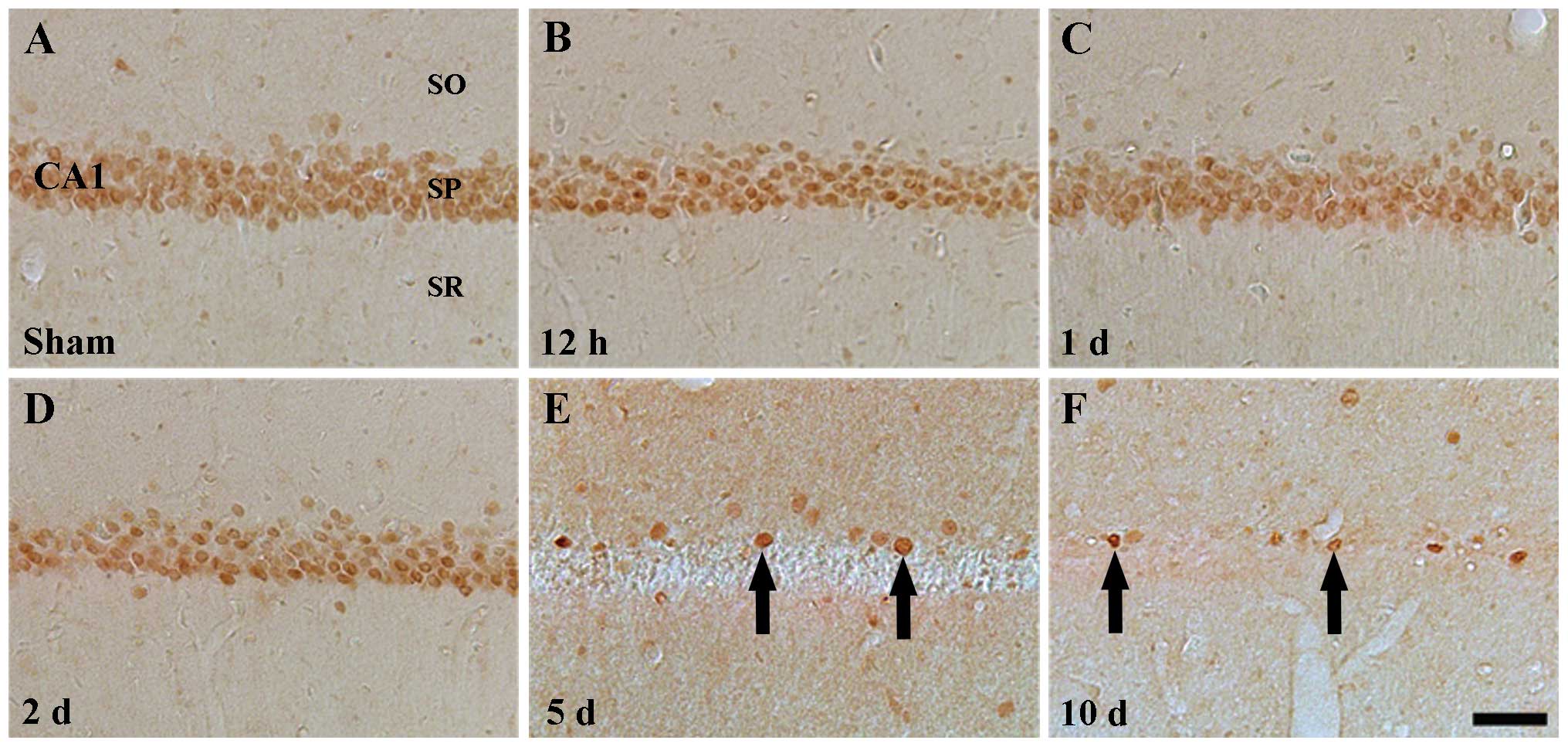
ID1 immunoreactivity
In the sham-operated group, ID1 immunoreactivity was
detected in the pyramidal neurons of the stratum pyramidale in the
hippocampal regions CA1–3, especially in the nucleus, but not in
the cytoplasm, of the pyramidal neurons (Table II, Figs. 2A and 3A). ID1 immunoreactivity was prominently
changed in the pyramidal cells of the CA1 region, and not of the
CA2/3 region, following I-R. In the CA1 region of the ischemia
groups, positive ID1 immunoreactivity was detected until 2 days
post-ischemia (Table II, Fig. 2B–D). Five days following I-R, ID1
immunoreactivity was hardly detectable in the pyramidal neurons of
the stratum pyramidale of the CA1 region (Table II, Fig. 2E) and following this time point,
the pattern of ID1 immunoreactivity in the CA1 region was similar
to that observed 5 days following I-R (Table II, Fig. 2F). At these time points (5 and 10
days), a few ID1+ cells were observed near the stratum
pyramidale (Fig. 2E and F).
| Table IISemi-quantitative analysis of ID1 and
ID4 immunoreactivity in cells of the Cornu Ammonis regions
CA1 and CA3 of the sham-operated and ischemia groups. |
Table II
Semi-quantitative analysis of ID1 and
ID4 immunoreactivity in cells of the Cornu Ammonis regions
CA1 and CA3 of the sham-operated and ischemia groups.
| | | Time after
ischemia/reperfusion |
|---|
| | |
|
|---|
| Antibody | Region | Cell type | Sham | 12 h | 1 d | 2 d | 5 d | 10 d |
|---|
| ID1 | CA1 | CSP | ++ | ++ | ++ | + | − | ± |
| ID1 | CA1 | CSOR | ± | ± | ± | ± | ++ | ++ |
| ID1 | CA3 | CSP | ++ | ++ | ++ | ++ | ++ | ++ |
| ID1 | CA3 | CSOR | ++ | ++ | ++ | ++ | ++ | ++ |
| ID4 | CA1 | CSP | ± | ± | ± | ± | ± | ± |
| ID4 | CA1 | CSOR | + | + | + | ++ | ++ | ++ |
| ID4 | CA3 | CSP | ± | ± | ± | ± | ± | ± |
| ID4 | CA3 | CSOR | + | + | + | + | + | + |
By contrast, ID1 immunoreactivity in the CA2/3
region of the sham-operated group was similar to that observed in
the CA1 region (Table II,
Fig. 3A). In the ischemia groups,
ID1 immunoreactivity was barely changed upon I-R (Table II, Fig. 3B–F).
ID2 and ID3 immunoreactivity
No ID2+ or ID3+ cells were
detected in the hippocampus proper (CA1-CA3) of both the
sham-operated and the ischemia groups (data not shown).
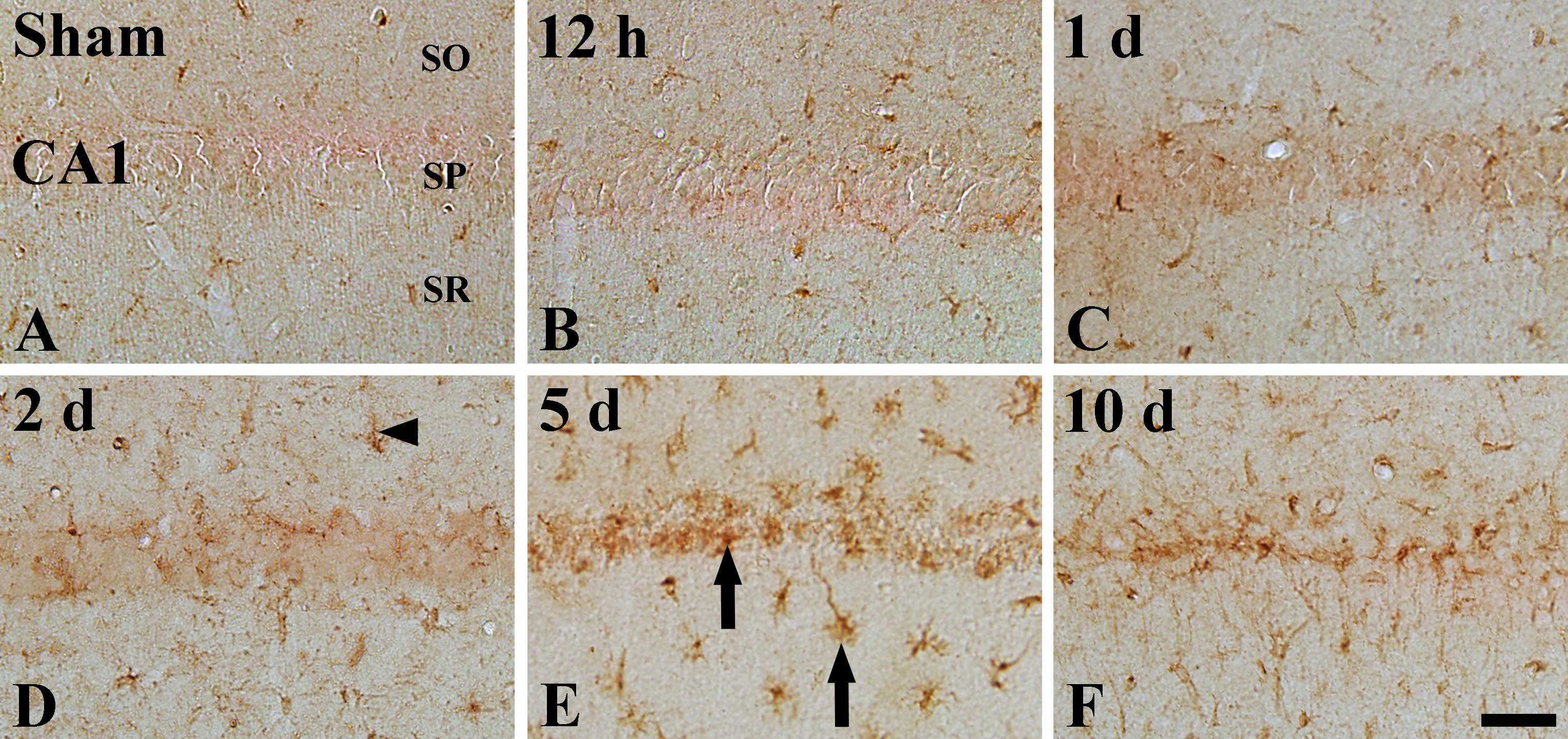
ID4 immunoreactivity
ID4 immunoreactivity in the sham-operated group was
weakly detected in the hippocampal CA1 region; ID4 immunoreactivity
was detected in cells that had short processes (indicated by
arrowhead in Fig. 3A), in the
stratum oriens and the stratum radiatum (Table II). In the the CA1 region of the
ischemia groups, ID4 immunoreactivity was not changed until 1 day
after I-R (Table II, Fig. 4B and C). However, ID4
immunoreactivity was slightly increased 2 days following I-R
(Table II, Fig. 4D) and reached its highest level 5
days following I-R (Table II,
Fig. 4E). Ten days after I-R, ID4
immunoreactivity was slightly decreased in the ischemic CA1 region,
and ID4+ processes were longer than those observed at 5
days post-ischemia (Table II,
Fig. 4F).
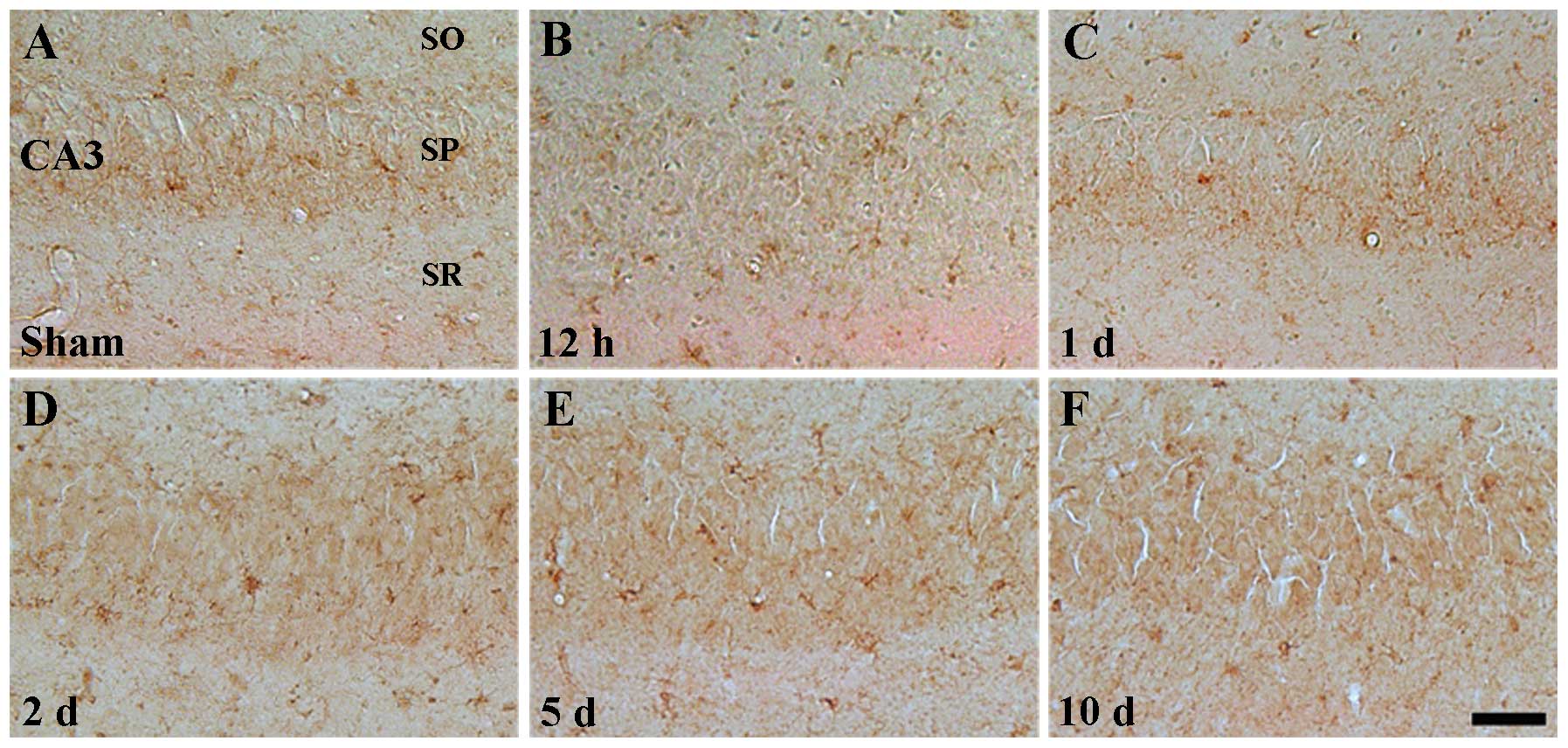
Weak ID4 immunoreactivity was also detected in the
CA3 region of the sham-operated group (Table II, Fig. 5A). In the ischemia groups, the
pattern of ID4 immunoreactivity was similar to that of the
sham-operated group (Table II,
Fig. 5B–F).
Colocalization of ID1/GAD67 and
ID4/Iba-1
ID1 and ID4 immunoreactivities were detected in
non-pyramidal cells of the CA1 region, but not in pyramidal
neurons, from 5 days post-I-R (Figs.
2E and 4E). In order to
identify the cell types of non-pyramidal ID1+ and
ID4+ cells, double immunofluorescence staining was
performed for ID1/GAD67, ID4/GFAP and ID4/Iba-1 in the hippocampal
CA1 region at 5 days post-ischemia. We found that ID1+
cells colocalized with GAD67+ GABAergic interneurons
(Fig. 6A–C). In addition,
ID4+ non-pyramidal cells colocalized with
Iba-1+ microglia, but not with GFAP+
astrocytes (data not shown), in the ischemic CA1 region (Fig. 6D–F).
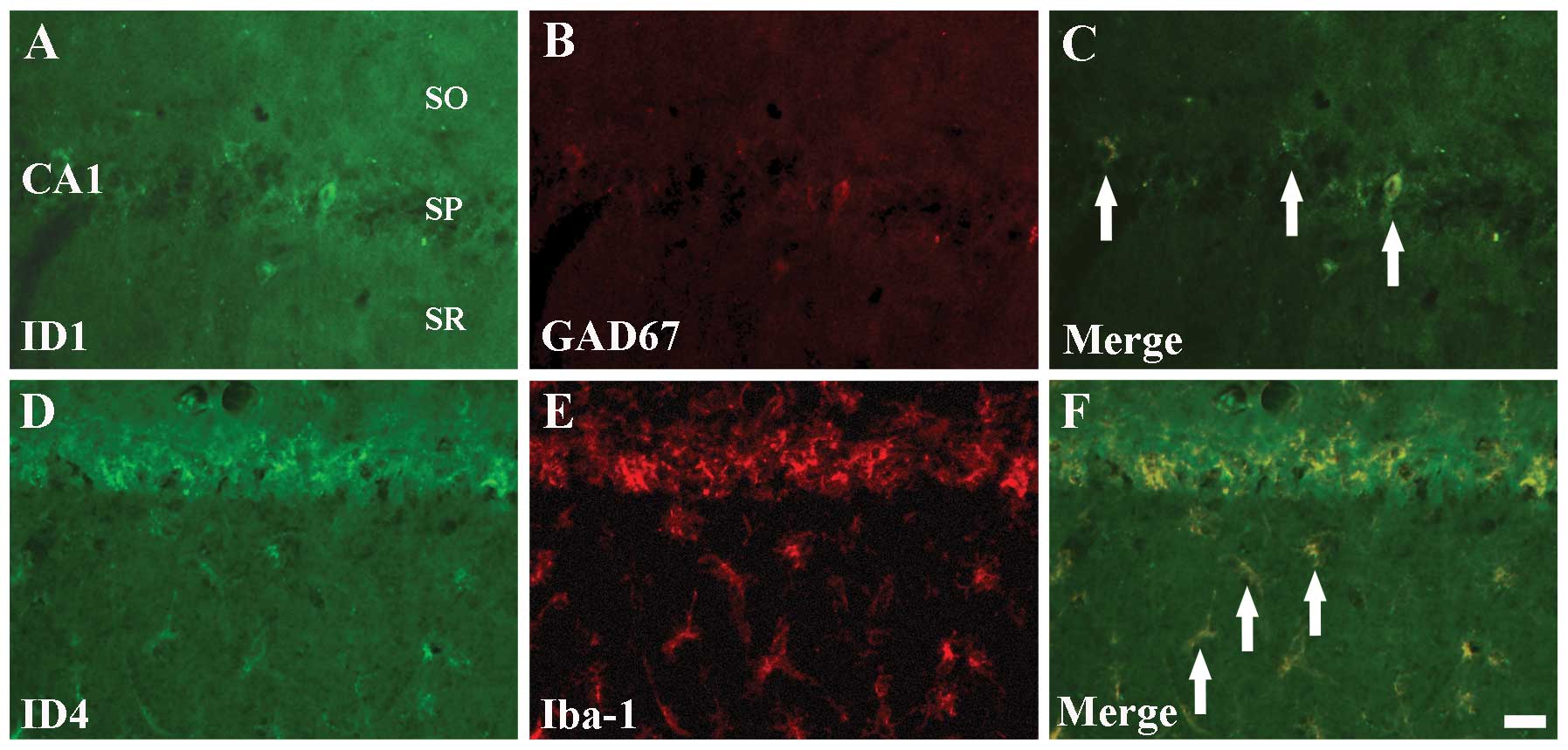 | Figure 6Double immunofluorescence staining
for (A) ID1 (green), (B) GAD67 (red), (C) ID1+GAD67 (merged image),
(D) ID4 (green), (E) Iba-1 (red) and (F) ID4+Iba-1 (merged image)
in the Cornu Ammonis region CA1 5 days after
ischemia-reperfusion. ID1+ cells colocalize with
GAD67+ GABAergic interneurons (C, arrows);
ID4+ cells colocalize with GFAP+ astrocytes
(F, arrows). ID, inhibitors of DNA binding/differentiation
proteins; GAD67, glutamic acid decarboxylase 67; GABA,
γ-aminobutyric-acid; Iba-1, ionized calcium-binding adapter
molecule 1; GFAP, glial fibrillary acidic protein; SO, stratum
oriens; SP, stratum pyramidale; and SR, stratum radiatum. Scale
bar=20 μm. |
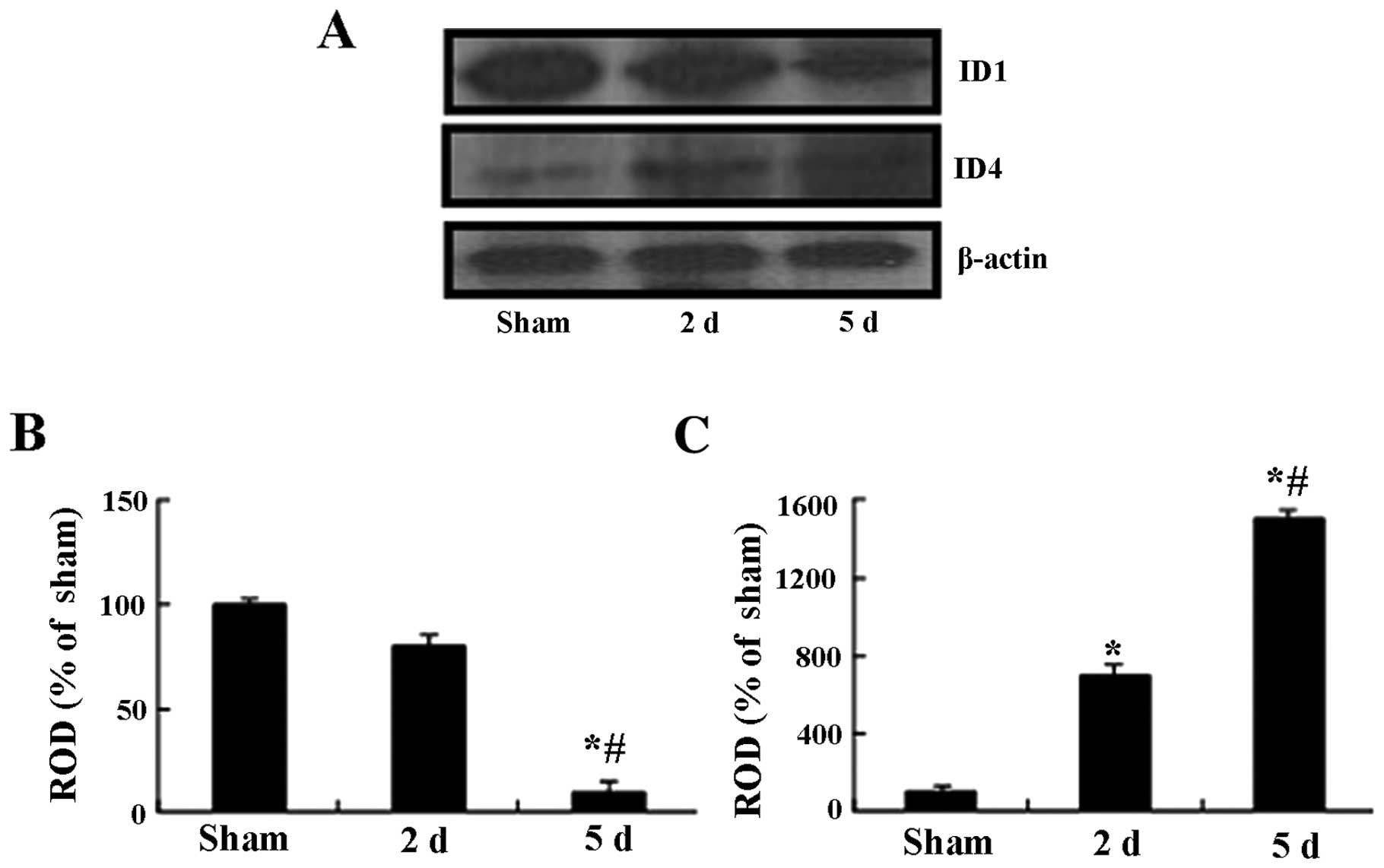
Changes in ID1 and ID4 protein
levels
Western blot analysis showed that the patterns of
changes in the ID1 and ID4 protein levels in the hippocampal CA1
region following I-R were similar to those observed in the
immunohistochemical data. In the ischemia groups, the ID1 protein
level was slightly decreased at 2 days following I-R compared to
that of the sham-operated group; however, the ID1 protein level was
significantly decreased 5 days following I-R compared to that of
the sham-operated group (Fig. 7).
The ID4 protein level was increased compared to the sham-operated
group at 2 days following I-R and peaked at 5 days post-I-R
(Fig. 7).
Discussion
In the present study, we observed neuronal damage in
the gerbil hippocampal CA1 region 5 days after I-R using NeuN
immunohistochemistry and F-J B histofluorescence staining. F-J B
has been used as a good fluorescent marker for the detection of
neuronal degeneration (26).
NeuN+ neurons were significantly decreased in the
stratum pyramidale of the CA1 region 5 days following I-R, and
numerous F-J B+ neurons were detected in the stratum
pyramidale. This result is consistent with previous studies
(1,29).
Mature neurons are terminally-differentiated,
post-mitotic cells that do not follow the conventional cell-cycle
process. However, a number of studies have shown that in certain
neurological disorders, mature neurons reenter the process of cell
cycle and undergo cell damage/death including apoptosis (30–32),
because they are terminally-differentiated cells that can not
re-enter the cell cycle (32,33).
There is some evidence that the process of neuronal re-entry into
the cell cycle is associated with pathological conditions such as
stroke (34,35) and cerebral hypoxia-ischemia
(36). ID proteins can inhibit the
expression of cyclin-dependent inhibitors such as p21 via
inhibition of bHLH factors (10,37).
Moreover, the inhibition of ID protein synthesis by antisense
oligonucleotides prevents the re-entry of G0-arrested fibroblasts
into the cell cycle (38,39). These reports strongly suggest that
ID proteins play a role in the G0-S phase transition of the cell
cycle. To the best of our knowledge, no studies of the changes in
immunoreactivity of ID proteins following ischemic damage have been
reported yet, and the roles of ID in the ischemic brain remain
unclear. The present study demosntrated a method by which to
maintain neurons in the quiescent G0 phase and to protect neurons
from damage caused by cerebral ischemia. In this study, we observed
changes in ID1 immunoreactivity in the CA1 region following I-R;
ID1 immunoreactivity in the pyramidal neurons was markedly
decreased from 5 days after I-R, and the changes in the ID1 protein
level were in accordance with ID1 immunoreactivity. This finding
indicates that ID1 protein may be related to the delay of
damage/death of the CA1 pyramidal neurons following I-R injury. It
is notable that ID1 immunoreactivity was not distinctively changed
in the CA3 region following I-R. The CA3 region is known as the
most tolerant area of the hippocampus to ischemia (1,29).
In addition, we observed that ID1+ cells that survived
near the stratum pyramidale 5 days following I-R were GABAergic
interneurons. This is the first study to show that ID1 is expressed
in the GABAergic interneurons of the CA1 region following transient
cerebral ischemia. It has been previously reported that most of
GABAergic interneurons in the CA1 region are resistant to transient
cerebral ischemia (40,41). Our findings indicate that the ID1
protein may be involved in the survival of GABAergic neurons
following an ischemic insult.
In the present study, we found that ID4
immunoreactivity is gradually increased in the microglia in the CA1
region, and peaks 5 days following I-R. This is also the first
study reporting expression of ID4 in the microglia, but not
astrocytes, in the CA1 region. It is well known that ID4 plays an
important role in proliferation and differentiation in a variety of
cell types, including neurons (42–44).
In addition, it was reported that ID4 protein enhances the
translation of mRNAs encoding pro-angiogenic cytokines, such as
interleukin 8 and growth-regulated oncogene-α, and that ID4
increases the angiogenic potential of cancer cells (45). Based on these studies, we suggest
that ID4 plays a role in proliferation of the microglia following
ischemic damage. We do not have a solid hypothesis to explain the
increased expression of ID4 in the microglia following I-R, but it
is well known that microglia are hyperactivated and hypertrophied
in all the layers of the CA1 region following transient cerebral
ischemia and before the occurence of delayed neuronal death in the
CA1 pyramidal neurons (46).
Furthermore, numerous studies have shown that the activation of
microglia is associated with the development of neuronal death
during ischemia (47–49); microglia secrete large amounts of
cytotoxic and inflammatory mediators (50–52),
and contribute to the elimination of deleterious debris, promotion
of tissue repair and return to tissue homeostasis (53–55).
Therefore, it can be postulated that ID4 plays an important role in
microglial proliferation in response to ischemic damage. Additional
studies are needed to elucidate the exact role of ID4 in the
microglia following transient cerebral ischemia.
In summary, immunoreactivity and the protein level
of ID1 protein were apparently altered in the CA1 pyramidal neurons
following transient cerebral ischemia, while ID4 immunoreactivity
was detected in the microglia, but not the astrocytes, in the CA1
region following I-R. These results indicate that changes in the
expression of ID1 and ID4 may be associated with delayed neuronal
death and glial activation in the CA1 region following ischemic
damage.
Acknowledgements
The authors would like to thank Mr. Seung Uk Lee for
his technical help in this study. This study was supported by the
National Research Foundation of Korea (NRF), funded by the Ministry
of Education, Science and Technology (no. 2010-0010580) and by a
Priority Research Centers Program grant (NRF-2009-0093812) through
the NRF, funded by the Ministry of Science, ICT and Future
Planning.
References
|
1
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin CS, Polsky K, Nadler JV and Crain BJ:
Selective neocortical and thalamic cell death in the gerbil after
transient ischemia. Neuroscience. 35:289–299. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pulsinelli WA, Brierley JB and Plum F:
Temporal profile of neuronal damage in a model of transient
forebrain ischemia. Ann Neurol. 11:491–498. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Won MH, Kang T, Park S, et al: The
alterations of N-methyl-D-aspartate receptor expressions and
oxidative DNA damage in the CA1 area at the early time after
ischemia-reperfusion insult. Neurosci Lett. 301:139–142. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rastogi L, Godbole MM, Ray M, et al:
Reduction in oxidative stress and cell death explains
hypothyroidism induced neuroprotection subsequent to
ischemia/reperfusion insult. Exp Neurol. 200:290–300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Candelario-Jalil E, Alvarez D, Merino N
and Leon OS: Delayed treatment with nimesulide reduces measures of
oxidative stress following global ischemic brain injury in gerbils.
Neurosci Res. 47:245–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kee Y and Bronner-Fraser M: To proliferate
or to die: role of Id3 in cell cycle progression and survival of
neural crest progenitors. Genes Dev. 19:744–755. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massari ME and Murre C: Helix-loop-helix
proteins: regulators of transcription in eucaryotic organisms. Mol
Cell Biol. 20:429–440. 2000. View Article : Google Scholar
|
|
9
|
Perk J, Iavarone A and Benezra R: Id
family of helix-loop-helix proteins in cancer. Nat Rev Cancer.
5:603–614. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ruzinova MB and Benezra R: Id proteins in
development, cell cycle and cancer. Trends Cell Biol. 13:410–418.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kremer D, Aktas O, Hartung HP and Kury P:
The complex world of oligodendroglial differentiation inhibitors.
Ann Neurol. 69:602–618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
13
|
Jen Y, Manova K and Benezra R: Each member
of the Id gene family exhibits a unique expression pattern in mouse
gastrulation and neurogenesis. Dev Dyn. 208:92–106. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neuman T, Keen A, Zuber MX, Kristjansson
GI, Gruss P and Nornes HO: Neuronal expression of regulatory
helix-loop-helix factor Id2 gene in mouse. Dev Biol. 160:186–195.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Riechmann V and Sablitzky F: Mutually
exclusive expression of two dominant-negative helix-loop-helix
(dnHLH) genes, Id4 and Id3, in the developing brain of the mouse
suggests distinct regulatory roles of these dnHLH proteins during
cellular proliferation and differentiation of the nervous system.
Cell Growth Differ. 6:837–843. 1995.PubMed/NCBI
|
|
16
|
Andres-Barquin PJ, Hernandez MC and Israel
MA: Id genes in nervous system development. Histol Histopathol.
15:603–618. 2000.PubMed/NCBI
|
|
17
|
Elliott RC, Khademi S, Pleasure SJ, Parent
JM and Lowenstein DH: Differential regulation of basic
helix-loop-helix mRNAs in the dentate gyrus following status
epilepticus. Neuroscience. 106:79–88. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rubenstein JL, Anderson S, Shi L,
Miyashita-Lin E, Bulfone A and Hevner R: Genetic control of
cortical regionalization and connectivity. Cereb Cortex. 9:524–532.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tzeng SF and de Vellis J: Id1, Id2 and Id3
gene expression in neural cells during development. Glia.
24:372–381. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fukuchi T, Katayama Y, Kamiya T, McKee A,
Kashiwagi F and Terashi A: The effect of duration of cerebral
ischemia on brain pyruvate dehydrogenase activity, energy
metabolites and blood flow during reperfusion in gerbil brain.
Brain Res. 792:59–65. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lorrio S, Negredo P, Roda JM, Garcia AG
and Lopez MG: Effects of memantine and galantamine given separately
or in association, on memory and hippocampal neuronal loss after
transient global cerebral ischemia in gerbils. Brain Res.
1254:128–137. 2009. View Article : Google Scholar
|
|
22
|
Zhang YB, Kan MY, Yang ZH, et al:
Neuroprotective effects of N-stearoyltyrosine on transient global
cerebral ischemia in gerbils. Brain Res. 1287:146–156. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Q, Sun AY, Pardeike J, Muller RH,
Simonyi A and Sun GY: Neuroprotective effects of a nanocrystal
formulation of sPLA2 inhibitor PX-18 in cerebral
ischemia/reperfusion in gerbils. Brain Res. 1285:188–195. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hwang IK, Eum WS, Yoo KY, et al: Copper
chaperone for Cu,Zn-SOD supplement potentiates the Cu,Zn-SOD
function of neuroprotective effects against ischemic neuronal
damage in the gerbil hippocampus. Free Radic Biol Med. 39:392–402.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park OK, Yoo KY, Lee CH, et al:
Arylalkylamine N-acetyltransferase (AANAT) is expressed in
astrocytes and melatonin treatment maintains AANAT in the gerbil
hippocampus induced by transient cerebral ischemia. J Neurol Sci.
294:7–17. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmued LC and Hopkins KJ: Fluoro-Jade B:
A high affinity fluorescent marker for the localization of neuronal
degeneration. Brain Res. 874:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loskota WJ, Lomax LP and Verity MA: A
stereotaxic atlas of the Mongolian gerbil brain (Meriones
unguiculatus). Loskota William James, Lomax Peter and Verity M
Anthony: Ann Arbor Science; Ann Arbor, MI: 1974
|
|
28
|
Lee CH, Park JH, Choi JH, Yoo KY, Ryu PD
and Won MH: Heat shock protein 90 and its cochaperone, p23, are
markedly increased in the aged gerbil hippocampus. Exp Gerontol.
46:768–772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee CH, Moon SM, Yoo KY, et al: Long-term
changes in neuronal degeneration and microglial activation in the
hippocampal CA1 region after experimental transient cerebral
ischemic damage. Brain Res. 1342:138–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen MD, Boudreau M, Kriz J,
Couillard-Despres S, Kaplan DR and Julien JP: Cell cycle regulators
in the neuronal death pathway of amyotrophic lateral sclerosis
caused by mutant superoxide dismutase 1. J Neurosci. 23:2131–2140.
2003.PubMed/NCBI
|
|
31
|
Vincent I, Rosado M and Davies P: Mitotic
mechanisms in Alzheimer’s disease? J Cell Biol. 132:413–425. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Byrnes KR and Faden AI: Role of cell cycle
proteins in CNS injury. Neurochem Res. 32:1799–1807. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rashidian J, Iyirhiaro GO and Park DS:
Cell cycle machinery and stroke. Biochim Biophys Acta.
1772:484–493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wen Y, Yang S, Liu R, Brun-Zinkernagel AM,
Koulen P and Simpkins JW: Transient cerebral ischemia induces
aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like
tauopathy in female rats. J Biol Chem. 279:22684–22692. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang F, Corbett D, Osuga H, et al:
Inhibition of cyclin-dependent kinases improves CA1 neuronal
survival and behavioral performance after global ischemia in the
rat. J Cereb Blood Flow Metab. 22:171–182. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yokota Y and Mori S: Role of Id family
proteins in growth control. J Cell Physiol. 190:21–28. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hara E, Yamaguchi T, Nojima H, et al:
Id-related genes encoding helix-loop-helix proteins are required
for G1 progression and are repressed in senescent human
fibroblasts. J Biol Chem. 269:2139–2145. 1994.PubMed/NCBI
|
|
39
|
Barone MV, Pepperkok R, Peverali FA and
Philipson L: Id proteins control growth induction in mammalian
cells. Proc Natl Acad Sci USA. 91:4985–4988. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fukuda T, Nakano S, Yoshiya I and
Hashimoto PH: Persistent degenerative state of non-pyramidal
neurons in the CA1 region of the gerbil hippocampus following
transient forebrain ischemia. Neuroscience. 53:23–38. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tortosa A and Ferrer I: Parvalbumin
immunoreactivity in the hippocampus of the gerbil after transient
forebrain ischaemia: a qualitative and quantitative sequential
study. Neuroscience. 55:33–43. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Benezra R, Davis RL, Lockshon D, Turner DL
and Weintraub H: The protein Id: a negative regulator of
helix-loop-helix DNA binding proteins. Cell. 61:49–59. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nagata Y and Todokoro K: Activation of
helix-loop-helix proteins Id1, Id2 and Id3 during neural
differentiation. Biochem Biophys Res Commun. 199:1355–1362. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lyden D, Young AZ, Zagzag D, et al: Id1
and Id3 are required for neurogenesis, angiogenesis and
vascularization of tumour xenografts. Nature. 401:670–677. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fontemaggi G, Dell’Orso S, Trisciuoglio D,
et al: The execution of the transcriptional axis mutant p53, E2F1
and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol.
16:1086–1093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sugawara T, Lewen A, Noshita N, Gasche Y
and Chan PH: Effects of global ischemia duration on neuronal,
astroglial, oligodendroglial and microglial reactions in the
vulnerable hippocampal CA1 subregion in rats. J Neurotrauma.
19:85–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hailer NP, Jarhult JD and Nitsch R:
Resting microglial cells in vitro: analysis of morphology and
adhesion molecule expression in organotypic hippocampal slice
cultures. Glia. 18:319–331. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hwang IK, Yoo KY, Kim DW, et al: Ionized
calcium-binding adapter molecule 1 immunoreactive cells change in
the gerbil hippocampal CA1 region after ischemia/reperfusion.
Neurochem Res. 31:957–965. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schwartz M, Butovsky O, Bruck W and
Hanisch UK: Microglial phenotype: is the commitment reversible?
Trends Neurosci. 29:68–74. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Colton CA and Gilbert DL: Production of
superoxide anions by a CNS macrophage, the microglia. FEBS Lett.
223:284–288. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Han HS, Qiao Y, Karabiyikoglu M, Giffard
RG and Yenari MA: Influence of mild hypothermia on inducible nitric
oxide synthase expression and reactive nitrogen production in
experimental stroke and inflammation. J Neurosci. 22:3921–3928.
2002.PubMed/NCBI
|
|
52
|
Suzuki S, Tanaka K, Nogawa S, et al:
Temporal profile and cellular localization of interleukin-6 protein
after focal cerebral ischemia in rats. J Cereb Blood Flow Metab.
19:1256–1262. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hashimoto M, Nitta A, Fukumitsu H, Nomoto
H, Shen L and Furukawa S: Involvement of glial cell line-derived
neurotrophic factor in activation processes of rodent macrophages.
J Neurosci Res. 79:476–487. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Laurenzi MA, Arcuri C, Rossi R, Marconi P
and Bocchini V: Effects of microenvironment on morphology and
function of the microglial cell line BV-2. Neurochem Res.
26:1209–1216. 2001. View Article : Google Scholar
|
|
55
|
Lu YZ, Lin CH, Cheng FC and Hsueh CM:
Molecular mechanisms responsible for microglia-derived protection
of Sprague-Dawley rat brain cells during in vitro ischemia.
Neurosci Lett. 373:159–164. 2005. View Article : Google Scholar
|