Introduction
Gliomas, the most common type of primary brain
tumor, are classified into four grades (I–IV) according to the
pathological and clinical criteria established by the World Health
Organization (1). These include
specific histological subtypes, the majority of which are
astrocytomas, oligodendrogliomas and ependymomas.
The majority of normal cells produce energy via the
oxidation of pyruvate in the mitochondria. Cancer cells metabolize
more glucose than their normal counterparts, which is achieved
predominantly via aerobic glycolysis in the cytosol, producing high
levels of lactate (2). This
phenomenon is known as the Warburg effect. The persistent
activation of aerobic glycolysis in cancer cells can promote the
progression of cancer (3) and
mutations in metabolic enzymes can predispose cells to neoplasia,
either by activating oncogenes or by eliminating tumor-suppressor
genes (4).
The human genome has five isocitrate dehydrogenase
(IDH) genes encoding three distinct IDH enzymes, the
activities of which are dependent on either nicotinamide adenine
dinucleotide phosphate or nicotinamide adenine dinucleotide
(5). IDH enzymes catalyze the
oxidative decarboxylation of isocitrate to produce α-ketoglutarate
(α-KG), thus, IDH is involved in the metabolism and energy
production required for cell survival (6). Mutations in the IDH1 gene are
detected in >70% of secondary glioblastomas and lower-grade
gliomas (grades II–III) (7). The
predominant IDH1 mutation in glioma involves an amino acid
substitution at arginine 132 (IDH1R132), which resides
in the enzyme’s active site (8–10).
This mutation causes IDH1 to lose its normal catalytic activity and
gain the ability to catalyze the reduction of α-KG to produce
2-hydroxyglutarate (2-HG), leading to the accumulation of 2-HG and
altering cell metabolism (4,11).
As 2-HG is a competitive inhibitor of multiple α-KG-dependent
dioxygenases, this results in genome-wide changes in histone and
DNA methylation (12), which are
associated with tumorigenesis (6).
Although IDH1R132H is the most common
IDH1 mutation, the role of IDH1R132H in glioma
remains to be fully elucidated. Clinical studies have revealed that
patients with gliomas containing IDH1 mutations have
increased survival rates (7,13),
which may be correlated to increased rates of response to
chemotherapy or radiotherapy (14,15).
The present study investigated the functional impact
of the IDH1R132H mutation by establishing a clonal A172
cell line overexpressing IDH1R132H, and evaluating its
association with aerobic glycolysis.
Materials and methods
Cell culture and inhibitors
The A172 glioma cells (American Type Culture
Collection, Rockville, MD, USA) were maintained in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine
serum (FBS; Invitrogen Life Technologies, Carlsbad, CA, USA), 4.5
g/L glucose and 100 U/ml penicillin/streptomycin (both from Sangon
Biotech Co., Ltd., Shanghai, China). The cells were dissociated
using enzyme-free cell dissociation solution (EMD Millipore,
Bedford, MA, USA) and cultured at 37°C in a humidified atmosphere
of 5% CO2. The YC-1 (Sigma-Aldrich, St. Louis, MO, USA)
and 2-deoxyglucose (2-DG; Sigma-Aldrich) inhibitors were dissolved
in dimethyl sulfoxide (DMSO; Sangon Biotech Co., Ltd.) and
sterilized water, respectively, prior to use.
Construct generation
The full-length human wild-type (WT) IDH1 coding
sequence was amplified from the HEK293T cells (Sangon Biotech Co.,
Ltd.). The following synthesized primers (Invitrogen Life
Technologies) were used to fuse the cDNA in-frame with a FLAG tag
at the N-terminus: Forward 5′-TTT CGT ACG ATG GAT TAC AAG GAC GAC
GAT GAC AAG TCC AAA AAA AT-3′ containing the MluI site and
reverse 5′-TTT ACG CGT GGT ATG AAC TTA AAG TTT GG-3′ containing the
BsiWI site. This was inserted into the MluI- and
BsiWI-linearized pHR-SIN vector (Open Biosystems,
Huntsville, AL, USA). The IDH1R132H mutation was
generated in the pHR-SIN-IDH1WT using a QuikChange
Site-Directed Mutagenesis kit according to the manufacturer’s
instructions (Stratagene, Santa Clara, CA, USA) with the following
primer sequences: 5′-R132H, 5′-ACC TAT CAT CAT AGG TCA TCA TGC TTA
TGG G-3′ and 3′-R132H, 5′-TGA CCT ATG ATG ATA GGT TTT ACC CAT CCA
C-3′.
Stable overexpression of the
IDH1WT and IDH1R132H constructs in the A172
cells
The HEK293T cells (4×106) were seeded
into 60 mm plates in DMEM cell culture medium with 100 U/ml
penicillin/streptomycin 1 day prior to transfection. The cells were
transfected with either 5.2 μg pHR-SIN-IDH1WT or
pHR-SIN-IDH1R132H, 2.36 μg pSPAX2 and 0.8 μg pMD2G
plasmids using Lipofectamine 2000 (Invitrogen Life Technologies) in
DMEM. After 6 h, the transfection media was replaced with DMEM cell
culture medium without penicillin/streptomycin and the lentiviral
particles were harvested 72 h post-transfection. The A172 glioma
cells (total number: 2×105) were plated in 60 mm plates
in DMEM with 10% phosphate-buffered saline (PBS) at a confluence of
20% 1 day prior to transduction. The cells were transduced by
adding 3 ml media containing viral particles and 6 μg/ml polybrene
(Sigma-Aldrich). After 16 h, the conditioned media was replaced
with DMEM containing 15% FBS.
Immunofluorescence
The cells (5×105) were seeded onto glass
coverslips for 24 h, fixed with 4% paraformaldehyde and
permeabilized using 0.4% Triton X-100 in PBS (all from Sangon
Biotech Co., Ltd.) for 10 min. Non-specific binding was blocked
using PBS containing 5% FBS and the cells were incubated with
primary antibodies against FLAG (mouse monoclonal anti-FLAG M2;
1:500; F3165; Sigma-Aldrich) or IDH1R132H (mouse
monoclonal anti-human; 1:500; DIA-H09; Dianova GmbH, Hamburg,
Germany) at 4°C overnight. As a negative control, cells were
treated with immunoglobulin G under the same conditions. The cells
were washed three times in PBS prior to incubation with the
respective Alexa Fluor 594-conjuated secondary antibody (goat
anti-mouse; 1:1,000; 8890; Cell Signaling Technology, Inc.,
Danvers, MA, USA). Following staining, the cells were imaged using
a DM2500 Leica microscope (Leica Microsystems, Inc., Buffalo Grove,
IL, USA).
Protein extraction and western
blotting
The transfected A172 cells were immediately placed
on ice and washed with ice-cold PBS. The total protein was prepared
using radioimmunoprecipitation lysis buffer containing a protease
inhibitor cocktail (1:1,000; 04693124001; Roche Diagnostics GmbH,
Mannheim, Germany) and phenylmethanesulfonylfluoride
(Sigma-Aldrich). The proteins were resolved on 8–12%
SDS-polyacrylamide gels and transferred onto nitrocellulose
membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA) by
electroblotting. The membranes were blocked using Tris-buffered
saline with 0.1% Tween-20 (TBST; Sangon Biotech Co., Ltd.)
containing 5% non-fat dry milk for 1 h and incubated at 4°C
overnight with their respective primary antibodies against IDH1
(rabbit monoclonal anti-human; 1:1,000; 8137; Cell Signaling
Technology, Inc.), IDH1R132H (mouse monoclonal
anti-human; 1:1,000; DIA-H09; Dianova GmbH) or FLAG (mouse
monoclonal anti-FLAG M2; 1:1,000; F3165; Sigma-Aldrich).
Immunolabeling was detected using enhanced chemiluminescence
reagents (Sigma-Aldrich) and visualized using an Amersham Imager
600 (GE Healthcare, Uppsala, Sweden). β-actin was used as a loading
control.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
The total RNA was isolated from the cells using
TRIzol reagent (Invitrogen Life Technologies), according to the
manufacturer’s instructions. Subsequently, 1 μg total RNA was used
as a template for RT in a Moloney Murine Leukemia Virus Reverse
Transcriptase reaction (Fermentas, Burlington, Ontario, Canada).
RT-qPCR was performed using SYBR Green Master mix (Applied
Biosystems Life Technologies, Carlsbad, CA, USA) and β-actin was
used as an internal control. The following primers were used:
Glucose transporter 1 (Glut1), forward 5′-CTTTGTGGCCTTCTTTGAAGT-3′
and reverse 5′-CCACACAGTTGCTCCACAT-3′; hexokinase 2 (HK2), forward
5′-GATTGTCCGTAACATTCTCATCGA-3′ and reverse
5′-TGTCTTGAGCCGCTCTGAGAT-3′ and β-actin, forward
5′-GGCGGCACCACCATGTACCCT-3′ and reverse
5′-AGGGGCCGGACTCGTCATACT-3′. The PCR thermal cycling conditions
were as follows: Cycling began with 2 min at 50°C and 10 min at
95°C. Thermal cycling proceeded with 40cycles of 95°C for 0.5 min
and 60°C for 2 min. All reactions were performed in the 7500 Fast
Real-Time PCR System (Applied Biosystems, Foster City, CA, USA).
The comparative Ct method was used to calculate the expression of
mRNA relative to that of β-actin.
3-(4,5-dimethyl-thiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT) cell proliferation assay
The cells (5×103) were seeded into
96-well plates and cultured for 24, 48, 72 or 96 h, respectively.
Following the incubation period, MTT was added to each well at a
final concentration of 5 mg/ml and the cells were incubated at 37°C
for a further 4 h. The reaction was terminated by adding 150 μL
DMSO and the absorbance was measured using a BioTek ELISA reader
(BioTek Instruments Inc., Winooski, VT, USA) at a wavelength of 490
nm. Each experiment was performed in triplicate.
Colony formation assay
Cells were trypsinized (trypsin from Sigma-Aldrich)
to generate a single-cell suspension and seeded into three parallel
60-mm dishes with 500 cells in each dish. Fourteen days following
seeding, the colonies were stained with 0.5% crystal violet (Sangon
Biotech Co., Ltd.). The number of colonies containing ≥50 cells was
determined, and the results were reported as a percentage of the
number of colonies in untreated cultures of each corresponding
clone.
Measurement of glucose uptake
The cells were subjected to serum starvation for 12
h prior to being cultured in DMEM containing 25 mM glucose. The
cells were washed three times with PBS and incubated for 3 h in
DMEM containing 1 mCi/ml 2-Deoxy-D-(1,2–3H) glucose (PerkinElmer,
Inc., Boston, MA, USA). Following incubation, the cells were washed
three times using ice-cold PBS and solubilized in 1% SDS. The
radioactivity of each aliquot was determined in a scintillation
counter (LS6500 Multipurpose Scintillation Counter; Beckman
Coulter, Fullerton, CA, USA). Each assay was performed in
triplicate.
Measurement of extracellular lactate
The cells (5×105) were seeded into 60 mm
dishes and incubated in DMEM with 10% FBS at 37°C overnight. The
media was replaced with DMEM without FBS and the cells were
incubated for 1 or 2 h. The supernatant was collected and the
lactate levels were quantified by colorimetric assay using a
Lactate Assay kit (BioVision, Inc., Milpitas, CA, USA), according
to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 5.0 software (GraphPad Software, Inc., San Diego, CA,
USA). Statistical significance was determined using Student’s
t-test. The data are expressed as the mean ± standard error of the
mean. P<0.05 was considered to indicate a statistically
significant difference.
Results
Stable expression of IDH1WT
and IDH1R132H constructs in the A172 glioma cells
To determine the effects of overexpression of human
glioma-associated IDH1 mutant proteins within the context of an
isogenic glioma cell, N-terminus FLAG-tagged IDH1WT,
IDH1R132H and control constructs were produced and
stably expressed in the A172 glioma cells (Fig. 1A). Western blot analysis using
anti-FLAG, anti-IDH1 and mutant-specific anti-IDH1R132H
antibodies confirmed the expression of their respective IDH1
proteins (Fig. 1B). Subsequent
immunofluorescence analysis revealed that the steady-state cellular
localization of the IDH1R132H mutant protein was
indistinguishable from that of the IDH1WT protein,
indicating that the mutation caused no significant alteration to
the targeting of IDH1 (Fig. 1C and
D). The high specificity of the IDH1R132H antibody
was also indicated.
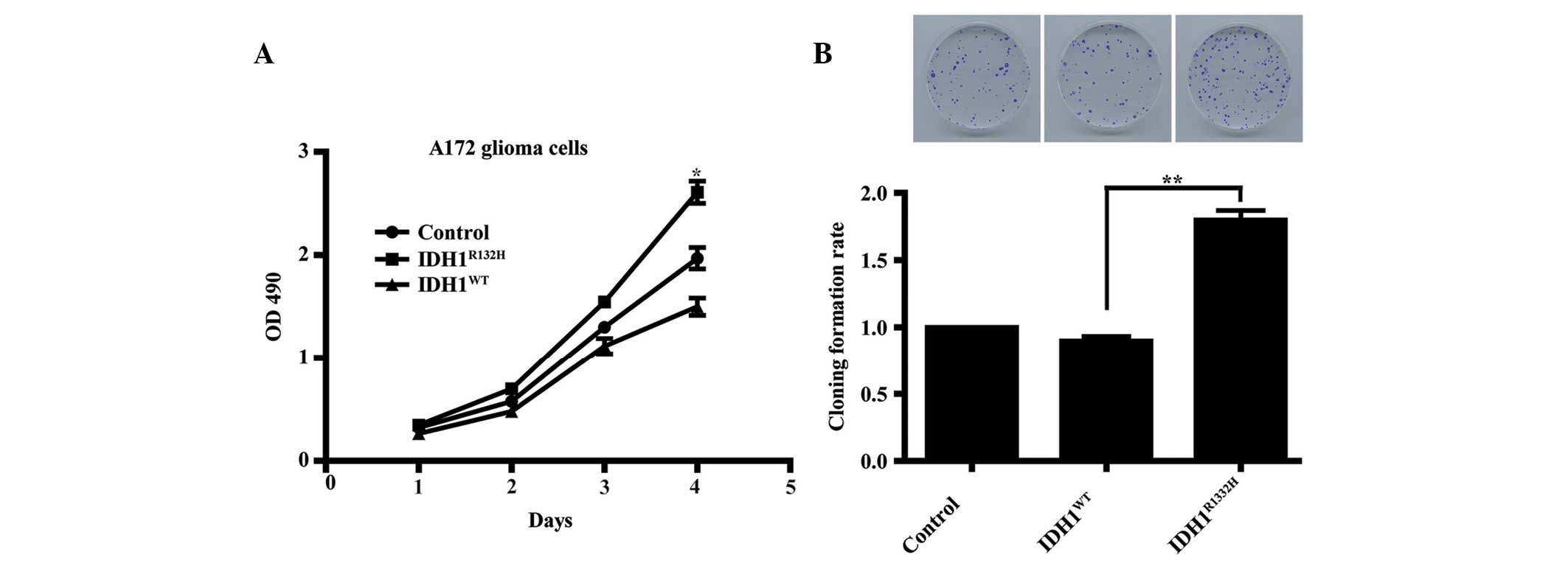
Overexpression of IDH1R132H
enhances A172 cell proliferation
The role of IDH1R132H in glioma remains
to be fully elucidated. To assess the importance of
IDH1R132H in A172 glioma cell growth, an MTT assay was
used to compare the proliferation rates between the two types of
IDH1-transfected cells and the control cells. The results revealed
that following incubation for 4 days, the growth curve for the
IDH1R132H cells were significantly higher compared with
those for the IDH1WT and the control cells (Fig. 2A; P<0.05). This observation was
supported by colony formation assays, which revealed that
clonogenicity was significantly increased in the mutant
IDH1R132H cells compared with the IDH1WT and
the control cells (Fig. 2B;
P<0.01).
IDH1R132H cells exhibit
increased levels of glycolysis
Increased glucose uptake and lactate secretion are
characteristics of proliferating cells undergoing glycolysis
(2,16). As IDH1 is important in metabolism,
the present study hypothesized that impaired IDH1 activity may
alter cell metabolism. Therefore, the levels of glucose uptake and
extracellular lactate were measured to investigate whether the
IDH1R132H mutation was responsible for a metabolic shift
in the IDH1R132H A172 cells. The results demonstrated
that the levels of glucose uptake and extracellular lactate in the
IDH1R132H cells were significantly higher compared with
the IDH1WT and control cells (Fig. 3A and B, respectively;
P<0.05).
 | Figure 3Overexpression of
IDH1R132H increases aerobic glycolysis in the A172
glioma cells. Quantification revealed that the rates of (A) glucose
uptake and (B) extracellular lactate secretion were significantly
increased in the IDH1R132H-expressing cells compared
with the IDH1WT and control cells. (C) Reverse
transcription quantitative polymerase chain reaction demonstrated
that the mRNA levels of Glut1 and HK2 were increased in the
IDH1R132H-expressing cells compared with the
IDH1WT and control cells (**P<0.01). (D)
Western blot analysis confirmed that the corresponding protein
levels of Glut1 and HK2 were also increased in the
IDH1R132H cells. (E) 3-(4,5-dimethyl-thiazol-2-yl)-2,
5-diphenyltetrazolium bromide assay revealed that the
IDHR132H-induced increase in cell proliferation was
inhibited by 2-DG, an inhibitor of glycolysis
(*P<0.05). The data are expressed as the mean ±
standard deviation based on three independent experiments. IDH1,
isocitrate dehydrogenase-1; WT, wild-type; R132H, mutation in
arginine 132; Glut1, glucose transporter 1; HK2, hexokinase 2;
2-DG, 2-deoxyglucose; OD, optical density. |
Glut1 and HK2 are important genes
involved in cell glycolysis (17–19),
therefore their expression levels were assessed in IDH1-transfected
cells using RT-qPCR and western blot analysis. The results revealed
that the mRNA and protein expression levels of Glut1 and HK2 were
increased significantly (Fig. 3C and
D; P<0.01) in the IDH1R132H cells compared with
the IDH1WT or control cells.
To elucidate whether the metabolic shift triggered
by IDH1R132H was involved in cell proliferation, the
A172 cells overexpressing IDH1 were treated with 2-DG, an inhibitor
of glycolysis, at a final concentration of 12 mM (20) and the effects on cell growth were
analyzed using an MTT assay. The results revealed that 2-DG
abrogated the IDH1R132H-induced cell proliferation
(Fig. 3E; P<0.05).
These findings suggested that
IDH1R132H-induced glycolysis promoted cell proliferation
in the A172 glioma cells.
Hypoxia inducible factor (HIF)-1α is
required for the enhanced glycolysis induced by
IDH1R132H
The overexpression of mutant IDH1 has been
demonstrated to reduce the levels of α-KG and increase the levels
of HIF-1α (11). Therefore, the
present study hypothesized that the IDH1R132H mutation
may be involved in promoting or stabilizing HIF-1α, thereby
enhancing aerobic glycolysis. Western blot analysis confirmed that
the protein expression of HIF-1α was increased in the
IDH1R132H cells compared with the IDH1WT and
the control cells (Fig. 4A).
To investigate whether HIF-1α is required in
IDH1R132H-induced glycolysis, the activity of HIF-1α was
inhibited in the IDH1-tranfected cells by treating the A172 cells
with YC-1 at a final concentration of 5 μM (21). Western blot analysis revealed that
the levels of Glut1 and HK2 were reduced in the
IDH1R132H cells following exposure to YC-1, whereas no
effects were observed in the IDH1WT or control cells
(Fig. 4B). Consistent with these
findings, YC-1 treatment significantly decreased the rate of
glucose uptake and extracellular lactate secretion in the mutant
IDH1R132H cells compared with the IDH1WT or
control cells (Fig. 4C and D,
respectively; P<0.05). These results demonstrated that HIF-1α
was important in modulating aerobic glycolysis in the
IDH1R132H A172 glioma cells.
Discussion
The association between the IDH1 mutation and
the development of glioma remains to be elucidated. The present
study demonstrated that the overexpression of IDH1R132H
increased cell proliferation in the A172 glioma cells via
glycolysis. Previous studies on the effect of the
IDH1R132H mutation in cell proliferation have been
contradictory (15,22). Zhu et al (22) reported that U87 cells stably
expressing IDH1R132H exhibit a higher proliferation rate
and degree of cell growth compared with wild-type U87 cells. By
contrast, Bralten et al (23) demonstrated that
U87MG-IDH1R132H cells exhibit decreased cell
proliferation and that mice injected with U87
IDH1R132H-expressing cells have significantly higher
survival rates compared with those injected with
IDH1WT-expressing cells. Based on results from the
present study, it was suggested that these conflicting effects may
be due to cell heterogeneity. In addition, the improved survival
rate of patients with the IDH1R132H mutant tumors may be
attributed to the enhanced sensitivity of IDH1R132H
glioma cells to radiation (15).
This suggests that an IDH1R132H mutation induced
alternative mechanism may be involved in tumor growth and its
response to therapy.
Compared with normal cells, glioma cells have a high
rate of aerobic glycolysis (24),
which is fundamental in cell proliferation (25,26).
To demonstrate that the IDH1R132H mutation leads to
enhanced aerobic glycolysis, thereby promoting cell proliferation
in glioma cells, the levels of glycolytic enzymes were measured.
The results demonstrated that the expression levels of Glut1 and
HK2 increased in the IDH1R132H cells and, furthermore,
the increase in cell proliferation was abrogated by the inhibition
of aerobic glycolysis, suggesting that IDH1R132H-induced
glycolysis was responsible for cell proliferation.
HIF-1α is important in the glycolytic metabolism of
cancer cells (27,28). Furthermore, Glut1 and
HK2 are target genes of HIF-1α (11,29,30).
The present study revealed that the protein expression of HIF-1α
was elevated in the IDH1R132H-expressing A172 cells and
inhibiting HIF-1α activity not only reduced the levels of Glut1 and
HK2, but also significantly decreased the rate of glucose uptake
and secretion of lactate in the mutant IDH1R132H cells.
This indicated that HIF-1α may act as an upstream signaling
molecule in the initiation of IDH1R132H-induced
glycolysis.
In conclusion, the results of the present study
suggested that the IDH1R132H mutation leads to increased
protein expression of HIF-1α, prompting a metabolic shift to
aerobic glycolysis via increase in the expression of glycolytic
enzymes, Glut1 and HK2, thereby enhancing glioma cell proliferation
in vitro. These results may provide insight into the
mechanisms underlying the development of glioma. Furthermore, by
identifying IDH1R132H as a potential chemotherapeutic
target, these findings may have broader implications in glioma
therapy, with potentially favorable outcomes in the treatment of
glioma from combined therapy involving the anti-HIF pathway.
Acknowledgments
The authors would like to thank Dr Dx Zhang (School
of Medicine, Shanghai Jiao Tong University, Shanghai, China) for
their technical support. This study was supported by the Shanghai
Science and Technology Committee (no. 13XD1402600), the Shanghai
Health and Family planning Commission (no. 2013SY024) and the State
Key Laboratory of Oncogenes and Related Genes (no. 90-14-01).
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, et al:
The 2007 WHO classification of tumours of the central nervous
system. Acta Neurophathol. 114:97–109. 2007. View Article : Google Scholar
|
|
2
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jang M, Kim SS and Lee J: Cancer cell
metabolism: implications for therapeutic targets. Exp Mol Med.
45:e452013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dang L, Jin S and Su SM: IDH mutations in
glioma and acute myeloid leukemia. Trends Mol Med. 16:387–397.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu Y, Huang R, Du J, Yang R, An N and
Liang A: Glioma-derived mutations in IDH: from mechanism to
potential therapy. Biochem Biophys Res Commun. 397:127–130. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Borodovsky A, Seltzer MJ and Riggins GJ:
Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2.
Curr Opin Oncol. 24:83–89. 2012. View Article : Google Scholar
|
|
7
|
Yan H, Parsons DW, Jin G, et al: IDH1 and
IDH2 mutations in gliomas. N Engl J Med. 360:765–773. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Turcan S, Rohle D, Goenka A, et al: IDH1
mutation is sufficient to establish the glioma hypermethylator
phenotype. Nature. 483:479–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kloosterhof NK, Bralten LB, Dubbink HJ,
French PJ and van den Bent MJ: Isocitrate dehydrogenase-1
mutations: a fundamentally new understanding of diffuse glioma?
Lancet Oncol. 12:83–91. 2011. View Article : Google Scholar
|
|
10
|
Gravendeel LA, Kloosterhof NK, Bralten LB,
et al: Segregation of non-p.R132H mutations in IDH1 in distinct
molecular subtypes of glioma. Hum Mutat. 31:E1186–E1199. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao S, Lin Y, Xu W, et al: Glioma-derived
mutations in IDH1 dominantly inhibit IDH1 catalytic activity and
induce HIF-1alpha. Science. 324:261–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu W, Yang H, Liu Y, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sanson M, Marie Y, Paris S, et al:
Isocitrate dehydrogenase 1 codon 132 mutation is an important
prognostic biomarker in gliomas. J Clin Oncol. 27:4150–4154. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Houillier C, Wang X, Kaloshi G, et al:
IDH1 or IDH2 mutations predict longer survival and response to
temozolomide in low-grade gliomas. Neurology. 75:1560–1566. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li S, Chou AP, Chen W, et al:
Overexpression of isocitrate dehydrogenase mutant proteins renders
glioma cells more sensitive to radiation. Neuro Oncol. 15:57–68.
2013. View Article : Google Scholar :
|
|
16
|
Elstrom RL, Bauer DE, Buzzai M, et al: Akt
stimulates aerobic glycolysis in cancer cells. Cancer Res.
64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mobasheri A, Richardson S, Mobasheri R,
Shakibaei M and Hoyland JA: Hypoxia inducible factor-1 and
facilitative glucose transporters GLUT1 and GLUT3: putative
molecular components of the oxygen and glucose sensing apparatus in
articular chondrocytes. Histol Histopathol. 20:1327–1338.
2005.PubMed/NCBI
|
|
18
|
Wolf A, Agnihotri S, Micallef J, et al:
Hexokinase 2 is a key mediator of aerobic glycolysis and promotes
tumor growth in human glioblastoma multiforme. J Exp Med.
208:313–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mueckler M, Caruso C, Baldwin SA, et al:
Sequence and structure of a human glucose transporter. Science.
229:941–945. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu H, Hu YP, Savaraj N, Priebe W and
Lampidis TJ: Hypersensitization of tumor cells to glycolytic
inhibitors. Biochemistry. 40:5542–5547. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chun YS, Yeo EJ, Choi E, et al: Inhibitory
effect of YC-1 on the hypoxic induction of erythropoietin and
vascular endothelial growth factor in Hep3B cells. Biochem
Pharmacol. 61:947–954. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu J, Cui G, Chen M, et al: Expression of
R132H mutational IDH1 in human U87 glioblastoma cells affects the
SREBP1a pathway and induces cellular proliferation. J Mol Neurosci.
50:165–171. 2013. View Article : Google Scholar
|
|
23
|
Bralten LB, Kloosterhof NK, Balvers R, et
al: IDH1 R132H decreases proliferation of glioma cell lines in
vitro and in vivo. Ann Neurol. 69:455–463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Griguer CE, Oliva CR and Gillespie GY:
Glucose metabolism heterogeneity in human and mouse malignant
glioma cell lines. J Neurooncol. 74:123–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lunt SY and Vander Heiden MG: Aerobic
glycolysis: meeting the metabolic requirements of cell
proliferation. Annu Rev Cell Dev Biol. 27:441–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: the metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marin-Hernandez A, Gallardo-Perez JC,
Ralph SJ, Rodriguez-Enriquez S and Moreno-Sanchez R: HIF-1alpha
modulates energy metabolism in cancer cells by inducing
over-expression of specific glycolytic isoforms. Mini Rev Med Chem.
9:1084–1101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Finley LW, Carracedo A, Lee J, et al:
SIRT3 opposes reprogramming of cancer cell metabolism through
HIF1alpha destabilization. Cancer Cell. 19:416–428. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dang CV, Le A and Gao P: MYC-induced
cancer cell energy metabolism and therapeutic opportunities. Clin
Cancer Res. 15:6479–6483. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mathupala SP, Rempel A and Pedersen PL:
Glucose catabolism in cancer cells: identification and
characterization of a marked activation response of the type II
hexokinase gene to hypoxic conditions. J Biol Chem.
276:43407–43412. 2001. View Article : Google Scholar : PubMed/NCBI
|