Introduction
Reperfusion is the most effective method of limiting
acute myocardial ischemia necrosis. However, reperfusion may also
be associated with a burst of reactive oxygen species (ROS)
production and intracellular calcium overload (1–3).
These dichotic effects result in paradoxical cardiocyte
dysfunction, a phenomenon termed myocardial ischemia reperfusion
injury (MIRI). The role of mitochondria during MIRI is critical, as
apoptosis is promoted by the mitochondrial pathway associated with
the mitochondrial permeability transition (MPT). MPT pores (MPTPs)
are located in the inner mitochondrial membrane, and when MIRI
induces cellular dysfunction, including increased Ca2+
concentrations and oxidative stress, mitochondria undergo swelling
and become uncoupled due to the opening of MPTPs (4). This leads to matrix swelling, release
of apoptotic signaling molecules and irreversible injury to the
mitochondria (5). The role of the
mitoKATP channel in modulating cardiac mitochondrial
function has been investigated previously (6,7).
Wakiyama et al (8)
confirmed that the opening of mitochondrial adenosine
triphosphate-sensitive potassium channels (mitoKATPs)
may reduce the release of cytochrome c, inhibit caspase-3
activation, stabilize the mitochondrial membrane potential (MMP)
and inhibit apoptosis (8).
Astragaloside is one of the most common Traditional
Chinese Medicines. It possesses multiple physiological and
pharmacological functions, including a protective effect in the
myocardium following ischemic injury. This effect may be associated
with clearing oxygen free radicals or reducing blood viscosity
(9,10). Notably, prior work has demonstrated
that Astragalus injection (Huangqizhusheye) may activate the
mitoKATPs and reduce MIRI (11). Astragaloside IV (AsIV) is an
extract of the monomer astragaloside (Fig. 1). AsIV produces various effects,
including protection against cerebral ischemia-reperfusion injury,
and the attenuation of renal tubulointerstitial fibrosis and
diabetes (12–14). Certain protective effects of AsIV
on cardiovascular disease have also been suggested (15–19).
In addition to providing cardioprotection during myocardial
ischemia (18,19), AsIV has also been demonstrated to
limit endothelial dysfunction induced by oxidative stress (15) and inhibit compensatory hypertrophy
of myocardial cells (16). The
results obtained in these previous studies provided the impetus to
study the mechanism of AsIV protection during reperfusion.
In the current study, the protective role of
mitoKATPs following AsIV treatment was investigated in
cardiocytes. Oxidative stress in cardiocytes was stimulated by
treating cultured cells with 0.2 mmol/l hydrogen peroxide
(H2O2) (20), resulting in calcium overload and
damage to the MMP. These three factors may act separately, or
interact with each other to cause damage to cellular structure,
function and metabolism, and ultimately result in cell death. This
type of injury simulates the pathogenesis of MIRI; thus, the
present study used this model to analyze the effect of AsIV on MIRI
and its underlying mechanisms.
Materials and methods
Cell culture and reagents
Primary rat neonatal cardiocytes were derived from
1-2-day-old Wistar rats (Experimental Animal Holding Facility of
Jilin University, China). Briefly, the hearts were removed under
aseptic conditions and the ventricles were homogenized in D-Hank’s
buffer. The tissue fragments were digested by stepwise exposure to
0.125% pancreatin (Gibco Life Technologies, Carlsbad, CA, USA). The
dissociated cells were pre-plated for 90 min to remove fibroblasts,
and the non-adherent cardiocytes were then plated at a density of
1×105 cells/mm2. Cells were cultured in
Iscove’s modified Dulbecco’s medium supplemented with 10% fetal
bovine serum and 1% penicillin/streptomycin (BHKT Clinical Reagent
Co., Ltd, Beijing, China), and maintained at 37°C in an incubator
with 95% O2 and 5% CO2. All animal studies
were performed in accordance with the Guide for the Care and Use of
Laboratory Animals and approved by the Institutional Animal Care
and Use Committee of Jilin University (Changchun, China).
2,7-Dichlorofluorescein diacetate (DCFH), Fluo-3/AM,
Rhodamine 123 and Annexin V were obtained from Sigma-Aldrich (St.
Louis, MO, USA). The mitoKATP inhibitor
5-hydroxydecanoate (5-HD) was also obtained from Sigma-Aldrich.
Peroxidation injury
To induce oxidative stress, cells were cultured in
serum-free medium for 12 h when the cells were semiconfluent and
beating synchronously. For the peroxidation challenge, the cells
were cultured in medium with 0.2 mmol/l H2O2
(Beijing Chemical Factory, Beijing, China) and maintained at 37°C
for 24 h.
Experimental design
The present study utilized five experimental groups
of cardiocytes as follows: i) The dimethyl sulfoxide (DMSO, Beijing
Chemical Factory, Beijing, China) group, maintained in 0.1% DMSO;
ii) the H2O2 group, treated with 0.2 mmol/l
H2O2 for 24 h; iii) the
AsIV+H2O2 group, pretreated with 30 mg/l AsIV
(provided by Academy of Chinese Medical Sciences of Jilin Province,
Changchun, China) for 30 min prior to H2O2
injury; iv) the AsIV+H2O2+5-HD group,
pretreated with 50 μmol/l 5-HD for 5 min prior to the AsIV
pretreatment; and v) the nicorandil (NIC; Changchun Dazheng
International Trade Group Pharmaceutical Co., Ltd, Changchun,
China)+H2O2 group, pretreated with 120 mg/l
NIC for 30 min prior to H2O2 injury. The
NIC+H2O2 group was used as a positive
control. For all AsIV treatments, a 0.1% AsIV stock solution was
dissolved in DMSO.
Cardiocyte viability and lactate
dehydrogenase (LDH) activity
Cardiocyte survival was assessed using an MTT assay.
Briefly, cells were incubated with 200 μl medium in 96-well
plates, 5 μl MTT stock solution (5 mg/ml) was then added to
each well and the cells were incubated for a further 4 h. Blue
formazan precipitate was produced from the cells by adding 100
μl DMSO and gently shaking, and the absorbance was measured
at 570 nm using an A-5082 ELISA reader (Chengdu Taimeng Technology
Co., Ltd., Chengdu, China). LDH release was measured using a GF-200
Semi Automatic Biochemical Analyzer (Shandong Gaomi Caihong
Instruments Co., Ltd., Gaomi, China).
Analysis of ROS production
Intracellular ROS levels were analyzed using a flow
cytometer with DCFH staining. DCFH was dissolved in ethanol
(Beijing Chemical Factory, Beijing, China) to produce a 1 mmol/l
stock solution. Cells were subsequently washed three times with
phosphate-buffered saline (PBS) prior to the addition of DCFH (10
μM). Following a 10-min incubation in the dark, the cells
were washed three times then resuspended in PBS. Fluorescence was
measured using a fluorescence microplate reader at 488/525 nm
(Gemini XPS; Shanghai Spectrum Instruments Co., Ltd., Shanghai,
China).
Apoptosis analysis
The non-adherent myocytes were digested by treatment
with 0.25% pancreatin for 10 min. The cells were then washed twice
with PBS, and the percentage of apoptotic cells was determined with
an Coulter Epics XL flow cytometer (Beckman Coulter, Brea, CA, USA)
after double staining with the Annexin V-FITC apoptosis detection
kit (Tianjin Sungene Biotech Co., Ltd. China). Annexin V was used
as an apoptosis indicator and propidium iodide (PI) as a necrosis
indicator. An+PI− represents viable apoptotic
cells in the lower right quadrant.
Cytosolic calcium concentration,
[Ca2+]i
Fluo-3 was used as a Ca2+ indicator
during fluorescence imaging to assess [Ca2+]i. Changes
in [Ca2+]i were analyzed using a Fluoview FV500 confocal
laser scanning microscope (Olympus Corporation, Tokyo, Japan).
Cells in 24-well plates were washed with HEPES buffer and treated
with Fluo-3/AM (10 μmol/l) for 40 min in the dark. Following
removal of Fluo-3/AM, the cells were washed three times with HEPES,
and fresh HEPES was added with the various drug treatments as
described. Baseline fluorescence in normal cells was recorded for 1
min, then H2O2 was added and the change in
fluorescence was measured for 5 min. The results were analyzed
using software which accompanied the confocal microscope.
Alterations in the levels of green fluorescence were used to
evaluate [Ca2+]i.
MMP (ΔΨm)
Fluorescence imaging of ΔΨm was conducted using
Rhodamine 123 as an indicator. Rhodamine 123 is a
mitochondria-selective dye, which in a reaction driven by ΔΨm in
normal polarized mitochondria, assembles into red
fluorescence-emitting dimers forming J-aggregates. Changes in MMP
were analyzed using the confocal microscope. Cells in 24-well
plates were treated with Rhodamine 123 (5 μg/ml) for 10 min
away from light. Following removal of Rhodamine 123, the cells were
washed with PBS. The level of Rhodamine 123 was measured and
analyzed with the Fluoview Viewer software, version 1.7a (Olympus
Corporation).
Statistics
Data are expressed as the mean ± standard error of
the mean. Statistical significance was calculated using one-way
analysis of variance with Newman-Keuls post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of AsIV on cardiocyte viability
and the change in LDH activity
Pretreatment with AsIV significantly increased
cardiocyte viability compared with that of the
H2O2 group (P<0.05), while pretreatment
with 5-HD resulted in reduced viability (P<0.05 vs. the AsIV
group) (Fig. 2). As indicated in
Fig. 3, cardiocyte damage
resulting from H2O2 exposure was reflected by
increased LDH release (P<0.01 vs. the DMSO control group); which
was reduced by AsIV pretreatment (P<0.01 vs. the
H2O2 group) and significantly increased in
the 5-HD pretreatment group (P<0.05 vs. the AsIV group). These
results indicated that AsIV may protect cardiocytes against
H2O2 injury, while the mitoKATP
inhibitor 5-HD may partially reverse this effect.
 | Figure 3LDH and ROS levels in rat myocardial
cells. (LDH, U/l; ROS, optical density525/a.u).
##P<0.01 vs. the DMSO group, **P<0.01
vs. the H2O2 group, ΔP<0.05 vs.
the AsIV+H2O2 group. LDH, lactate
dehydrogenase; ROS, reactive oxygen species; DMSO, dimethyl
sulfoxide; H2O2, hydrogen peroxide; AsIV,
astragaloside IV; 5-HD, 5-hydroxydecanoate; NIC, nicorandil. |
AsIV alters oxidative stress and
[Ca2+]i
Elevated levels of ROS may contribute to
ischemia-reperfusion injury. Therefore, the present study evaluated
whether AsIV-mediated cardioprotection may be partially attributed
to a reduction in oxidative stress. ROS generation was assessed by
DCFH fluorescence. The results indicated that
H2O2 induced significant ROS generation in
cardiocytes compared with the DMSO control group (P<0.01)
(Fig. 3), which was reduced
following AsIV treatment (P<0.05 vs. the
H2O2 group). This protective effect was
significantly attenuated by 5-HD pretreatment (P<0.05). These
results indicate that AsIV may protect cardiocytes against
H2O2 injury by reducing oxidative stress; and
this effect may be associated with the mitoKATP.
Ca2+ overload is a key contributor to the
mitochondrial permeability transition leading to
ischemia-reperfusion injury. As displayed in Fig. 4, AsIV-pretreated cells presented
significantly lower [Ca2+]i compared with that of the
H2O2 group (P<0.01). 5-HD pretreatment
prior to AsIV exposure resulted in significantly higher
[Ca2+]i compared with that of the AsIV group
(P<0.01). These results indicated that AsIV may relieve
Ca2+ overload, an effect which is also associated with
the mitoKATP.
Changes in MMP
MPTP opening may be critical for the transition from
reversible to irreversible myocardial ischemia-reperfusion injury.
Therefore, the present study investigated MPTP dynamics in
cardiocytes using Rhodamine 123. The MMP was (93.80±19.53) 30 min
subsequent to AsIV treatment, which was significantly higher than
that in the H2O2 group (P<0.01) (Fig. 5). 5-HD pretreatment prior to AsIV
led to an MMP that was significantly lower than that of the AsIV
group (P<0.01). These results demonstrated that AsIV may protect
cardiocytes by stabilizing the MMP, and this effect may be
associated with the mitoKATP.
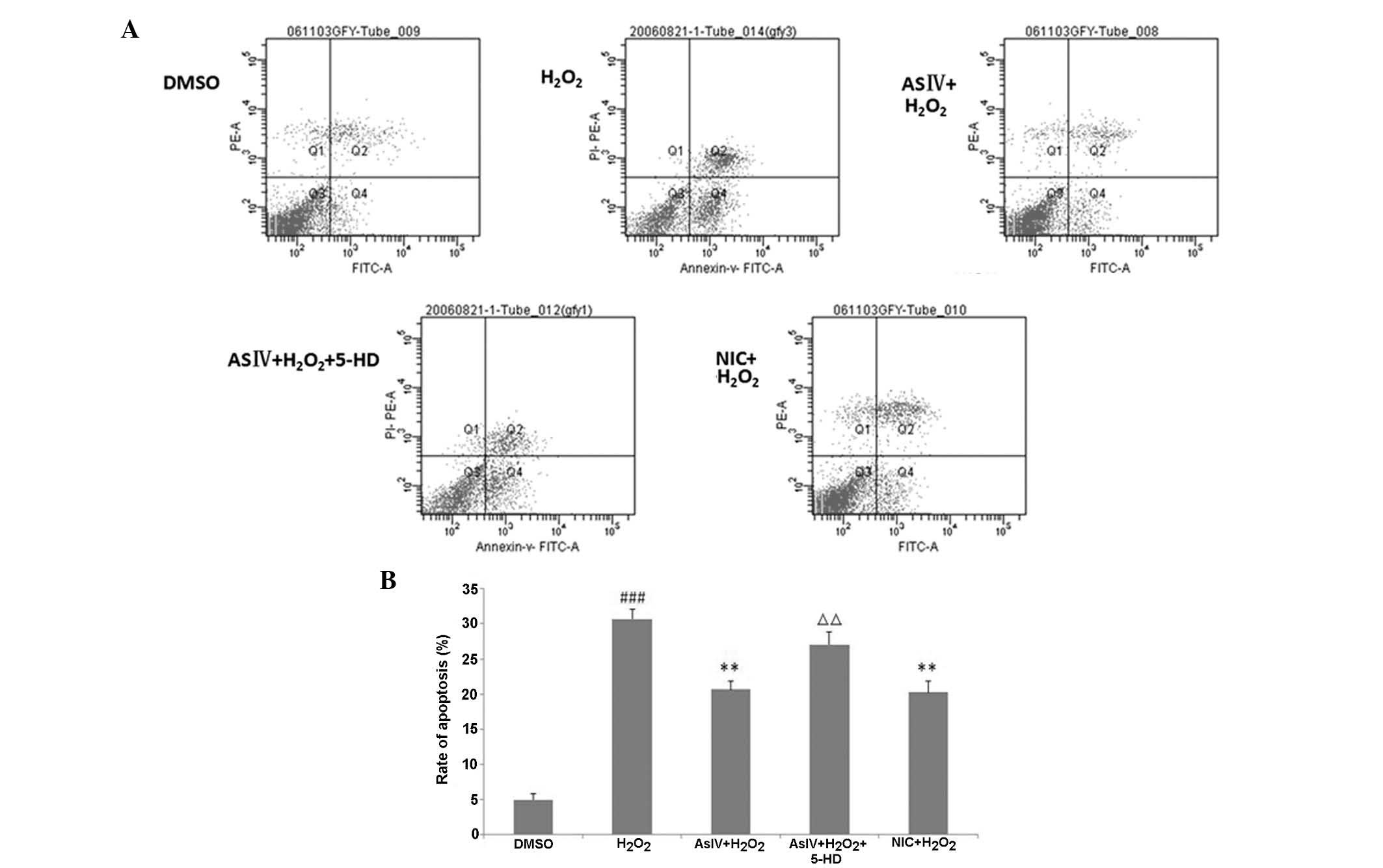
Changes in the rate of apoptosis
The effect of AsIV on cell apoptosis was analyzed by
measuring Annexin V positivity using flow cytometry. Pretreatment
with AsIV led to significantly reduced Annexin V-positivity
compared with the H2O2 group (P<0.01). By
contrast, pretreatment with 5-HD resulted in a significant increase
in the number of Annexin V-positive cells compared with the AsIV
group (P<0.01) (Fig. 6). These
results demonstrated that AsIV treatment may reduce apoptosis, and
this may be associated with the mitoKATP.
Discussion
The generation of ROS leads to damage to the
cellular membrane, and is able to evoke calcium overload by raising
membrane permeability, resulting in an inflow of calcium ions.
Calcium overload may activate calcium-dependent proteinase, which
catalyzes the conversion of xanthine dehydrogenase to xanthine
oxidase (XO). XO promotes xanthine decomposition into uric acid
with concomitant excess oxygen free radical generation.
Mitochondrial Ca2+ overload, coupled with a high
intracytoplasmic ROS burden, promotes the opening of the MPTPs,
which induces the release of calcium from mitochondria resulting in
mitochondrial swelling or failure. The combination of increased
ROS, Ca2+ overload and mitochondrial injury is a notable
cause of cardiocyte apoptosis.
The results of the current study demonstrated that
AsIV increased cardiocyte viability and reduced LDH release.
Additionally, pretreatment with AsIV significantly reduced
apoptosis, which demonstrated that AsIV was able to protect
cardiocytes from H2O2 injury and inhibit
apoptosis.
AsIV-mediated attenuation of ROS generation and
Ca2+ overload in H2O2-damaged
cardiocytes demonstrated that the protective effects of AsIV are
dependent on antioxidant activity and reducing the intracellular
calcium overload. These effects may protect cardiocytes by
attenuating damage to mitochondrial function.
Mitochondrial activity is required for efficient
function of the cardiovascular system. In response to
cardiovascular injury, mitochondrial dysfunction may result in
apoptosis and necrosis (21). A
change in mitochondrial permeability is considered the primary
event in the cell apoptosis cascade (22–24).
The fluctuations in MMP result in cytochrome c release,
which can initiate caspase-9 and -3 activation, and subsequent
apoptosis (25). In the present
study, AsIV was observed to protect against cell death by
modulating mitochondrial membrane permeability, which may attenuate
the release of cytochrome c and reduce cardiocyte apoptosis.
MitoKATP channels are located in the mitochondrial inner
membrane, and the opening of these channels has been suggested to
be protective against excessive mitochondrial Ca2+
accumulation, as mitoKATP opening leads to
depolarization, which reduces the driving force responsible for
mitochondrial Ca2+ uptake (26). MitoKATP opening also
regulates ROS generation; in early ischemia (30 min),
mitoKATP opening can increase the production of ROS to
begin cardiocyte protection by preconditioning. However, during
late ischemia (24 h) or reperfusion, mitoKATP opening
may reduce the production of ROS to reduce injury (27). Previous studies have indicated that
mito-KATP serves an important function in the protection
against ischemia-reperfusion injury by the mitoKATP
opening agent, diazoxide, using an siRNA method and
mitoKATP blocker 5-HD (28,29).
Wakiyama et al (8)
confirmed that mitoKATP opening may attenuate the
release of cytochrome c, inhibit caspase-3 release,
stabilize the MMP and inhibit apoptosis. Soeding et al
(30) also demonstrated that
levosimendan, a calcium sensitizer, preserves the contractile
responsiveness of hypoxic human myocardium via mitoKATPs
and potential pERK1/2 activation.
To investigate the link between AsIV and the
mitoKATP, the present study examined the effect of 5-HD
on the effects of AsIV. It was observed that pretreatment with 50
μmol/l 5-HD, (a specific mitoKATP blocker)
partially abrogated the protective effect of AsIV in the
cardiocytes. 5-HD led to a reduction in viability and MMP, an
increase in the levels of LDH and apoptosis and an increase in
calcium overload, compared with the AsIV group. These data indicate
that AsIV may protect cardiocytes from H2O2
injury through activating mitoKATP. This concentration
of 5-HD had no effect on cardiocytes alone (data not shown). It was
also observed that AsIV produced a similar protective effect to the
drug nicorandil, a mitoKATP activator.
Based on these findings, it was concluded that the
opening of mitoKATPs may be the principal mechanism
through which AsIV protects cardiocytes from
H2O2 injury. AsIV may inhibit apoptosis in
cardiocytes via several mechanisms: i) AsIV inhibits the initiation
of apoptosis by reducing the formation of ROS and intracellular
calcium overload; ii) AsIV inhibits the opening of MPTPs, which may
reduce the release of apoptosis-inducing proteins, including
cytochrome c, Smac and AIF; and iii) mitoKATP
opening may facilitate the signal transduction required for
protection by AsIV. Further studies are required to elucidate
whether other trigger factors underlie the protective effects of
AsIV. Additionally, the detailed downstream mediators of
mitoKATP require clarification.
Acknowledgments
This study was supported by the Science and
Technology Department of Jilin Province (grant no. YYZX201260).
References
|
1
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paradies G, Petrosillo G, Pistolese M, Di
Venosa N, Federici A and Ruggiero FM: Decrease in mitochondrial
complex I activity in ischemic/reperfused rat heart: involvement of
reactive oxygen species and cardiolipin. Circ Res. 94:53–59. 2004.
View Article : Google Scholar
|
|
3
|
Lemasters JJ, Theruvath TP, Zhong Z and
Nieminen AL: Mitochondrial calcium and the permeability transition
in cell death. Biochim Biophys Acta. 1787:1395–1401. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baines CP: The mitochondrial permeability
transition pore and ischemia-reperfusion injury. Basic Res Cardiol.
104:181–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weiss JN, Korge P, Honda HM and Ping P:
Role of the mitochondrial permeability transition in myocardial
disease. Circ Res. 93:292–301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fryer RM, Eells JT, Hsu AK, Henry MM and
Gross GJ: Ischemic preconditioning in rats: role of mitochondrial
K(ATP) channel in preservation of mitochondrial function. Am J
Physiol Heart Circ Physiol. 278:H305–H312. 2000.PubMed/NCBI
|
|
7
|
Garlid KD and Halestrap AP: The
mitochondrial K(ATP) channel-fact or fiction? J Mol Cell Cardiol.
52:578–583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wakiyama H, Cowan DB, Toyoda Y, Federman
M, Levitsky S and McCully JD: Selective opening of mitochondrial
ATP-sensitive potassium channels during surgically induced
myocardial ischemia decreases necrosis and apoptosis. Eur J
Cardiothorac Surg. 21:424–433. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou JY, Fan Y, Kong JL, Wu DZ and Hu ZB:
Effects of components isolated from Astragalus membranaceus Bunge
on cardiac function injured by myocardial ischemia reperfusion in
rats. Zhongguo Zhong Yao Za Zhi. 300–302. 2000.In Chinese.
|
|
10
|
Xu YC, Lan SL and Chen JY: Effects of
huangqi sijun decoction on thyroxin and cyclic nucleotide levels in
rat models with spleen deficiency syndrome. Chin Drug Res Clin
Pharm. 18:291–293. 2007.
|
|
11
|
Guan FY and Yu XX: The protective effect
of Astragalus membranaceus injection preconditioning on
experimental myocardial ischemia/reperfusion injury in rats. Chin J
Geront. 30:3126–3129. 2010.
|
|
12
|
Yang J, Li J, Lu J, Zhang Y, Zhu Z and Wan
H: Synergistic protective effect of astragaloside
IV-tetramethylpyrazine against cerebral ischemic-reperfusion injury
induced by transient focal ischemia. J Ethnopharmacol. 140:64–72.
2012. View Article : Google Scholar
|
|
13
|
Meng LQ, Tang JW, Wang Y, et al:
Astragaloside IV synergizes with ferulic acid to inhibit renal
tubulointerstitial fibrosis in rats with obstructive nephropathy.
Br J Pharmacol. 162:1805–1818. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xie W and Du L: Diabetes is an
inflammatory disease: evidence from traditional Chinese medicines.
Diabetes Obes Metab. 13:289–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiu LH, Xie XJ and Zhang BQ: Astragaloside
IV improves homocysteine-induced acute phase endothelial
dysfunction via antioxidation. Biol Pharm Bull. 33:641–646. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao Z, Wang W, Wang F, et al: Effects of
Astragaloside IV on heart failure in rats. Chin Med. 4:62009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guan FY, Li H, Sun W and Yang SJ:
Protective effects of Astragaloside IV on hydrogen peroxide-induced
injury of cultured neonatal rat myocardial cells. J Jilin Uni Med
Ed. 33:211–214. 2007.
|
|
18
|
Zhang WD, Chen H, Zhang C, Liu RH, Li HL
and Chen HZ: Astragaloside IV from Astragalus membranaceus shows
cardio-protection during myocardial ischemia in vivo and in vitro.
Planta Med. 72:4–8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu XL, Chen XJ, Ji H, et al: Astragaloside
IV improved intracellular calcium handling in hypoxia-reoxygenated
cardiocytes via the sarcoplasmic reticulum Ca-ATPase. Pharmacology.
81:325–332. 2008. View Article : Google Scholar
|
|
20
|
Yu WP, Xu GL, Shen CX and Qian ZY: Effects
of crocetin on the apoptosis and the changes of its related
regulating proteins caspase-3 and Bcl-2 induced by
H2O2 in myocardial cell. Chin J Pathoph.
22:54–57. 2006.
|
|
21
|
Smith MA and Schnellmann RG: Calpains,
mitochondria and apoptosis. Cardiovasc Res. 96:32–37. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grivicich I, Regner A, da Rocha AB, et al:
Irinotecan/5-fluorouracil combination induces alterations in
mitochondrial membrane potential and caspases on colon cancer cell
lines. Oncol Res. 15:385–392. 2005.
|
|
23
|
Kallio A, Zheng A, Dahllund J, Heiskanen
KM and Harkonen P: Role of mitochondria in tamoxifen-induced rapid
death of MCF-7 breast cancer cells. Apoptosis. 10:1395–1410. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shrivastava A, Tiwari M, Sinha RA, et al:
Molecular iodine induces caspase-independent apoptosis in human
breast carcinoma cells involving the mitochondria-mediated pathway.
J Biol Chem. 281:19762–19771. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi LG, Zhang GP and Jin HM: Inhibition of
microvascular endothelial cell apoptosis by angiopoietin-1 and the
involvement of cytochrome C. Chin Med J (Engl). 119:725–730.
2006.
|
|
26
|
Murata M, Akao M, O’Rourke B and Marban E:
Mitochondrial ATP-sensitive potassium channels attenuate matrix
Ca(2+) overload during simulated ischemia and reperfusion: possible
mechanism of cardioprotection. Circ Res. 89:891–898. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shahid M, Tauseef M, Sharma KK and Fahim
M: Brief femoral artery ischaemia provides protection against
myocardial ischaemia-reperfusion injury in rats: the possible
mechanisms. Exp Physiol. 93:954–968. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Q, Bie P, Tang C and Zhang YJ: An
experiment study on the role of bcl-2 in attenuation
ischemia-reperfusion injury in liver graft in mice induced by
mitoKATP channel opener diazoxide. J Clin Med Practi. 12:35–40.
2008.
|
|
29
|
Tratsiakovich Y, Gonon AT, Krook A, et al:
Arginase inhibition reduces infarct size via nitric oxide, protein
kinase C epsilon and mitochondrial ATP-dependent K+
channels. Eur J Pharmacol. 712:16–21. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soeding PF, Crack PJ, Wright CE, Angus JA
and Royse CF: Levosimendan preserves the contractile responsiveness
of hypoxic human myocardium via mitochondrial K(ATP) channel and
potential pERK 1/2 activation. Eur J Pharmacol. 655:59–66. 2011.
View Article : Google Scholar : PubMed/NCBI
|