Introduction
To date, doxorubicin (DOX) remains one of the most
widely administered anticancer therapeutic agents, due to its
potent therapeutic effects on cancer, including types of leukemia
and lymphoma, and breast cancer (1). However, its clinical application is
limited due to its marked toxic side-effects on the heart, which
may lead to dilated cardiomyopathy and congestive heart failure
(2). Numerous studies have
implicated reactive oxygen species (ROS) generation in the
cardiotoxicity associated with DOX, which ultimately results in
cardiomyocyte apoptosis (3,4).
However, the signal transduction pathway that links DOX-induced
oxidative stress and cardiac injuries remains to be fully
elucidated.
Hydrogen sulfide (H2S), a well-known
toxic gas, is regarded as the third gasotransmitter, along with
nitric oxide and carbon monoxide (5). Increasing evidence indicates that
H2S is significant in physiologic and pathophysiological
regulation of cardiovascular function (6). Our previous study revealed that
increased endogenous H2S generation in the early
reperfusion phase is important in ischemia preconditioning
(IPC)-elicited protection in isolated hearts (6). Furthermore, previous studies
demonstrated that extracellular signal-regulated protein kinase
(ERK) 1/2 is activated by oxidative stress and is hypothesized to
participate in cardiomyocyte apoptosis, as well as cardiac
pathologies (7,8).
Previous studies indicate that ERK1/2 may be
involved in DOX-induced cardiomyocyte injury. Lou et al
(9) reported that DOX caused an
early increase of ERK1/2 phosphorylation in the rat heart, which
was followed by the progressive decline of phosphorylated
(p)-ERK1/2 to the control three weeks after the final injection of
DOX. Liu et al (10)
observed that the ERKs/p53 signal transduction pathway is involved
in DOX-induced apoptosis in H9c2 cardiac myocytes. In addition,
H2S has been demonstrated to exert bidirectional effects
on ERK1/2 (11,12). H2S enhances activation
of ERK1/2 in mouse pancreatic acinar cells (13); however, it inhibits ERK1/2
activation in INS-1E insulin-secreting β-cell line cells (14). However, whether ROS-activated
ERK1/2 is involved in H2S protection against DOX-induced
cardiomyocyte injury remains unknown. These previous studies
provide a foundation upon which to investigate the role of
ROS-activated ERK1/2 in the protective effects of H2S
against DOX-induced cardiomyocyte injuries.
Thus, in the current study, H9c2 cardiac myocytes
were treated with 5 µM DOX to establish a
chemotherapy-induced cardiotoxicity model (15). Whether DOX induces activation of
ERK1/2 in H9c2 cardiac myocytes was investigated and the role of
ROS-activated ERK1/2 in the protective effect of H2S was
elucidated.
Materials and methods
Materials
MTT, Hoechst 33258, 2′,7′-dichlorofluorescein
diacetate (DCFH-DA), DOX, U0126, sodium hydrosulfide (NaHS), and
N-acetyl-L-cysteine (NAC) and H2O2 were
purchased from Sigma-Aldrich (St. Louis, MO, USA). All cell culture
medium components were purchased from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA) unless otherwise noted. The H9c2 cardiac
myocytes were obtained from the Shanghai Cell Library of China
(Shanghai, China; http://www.cellbank.org.cn/; originally purchased from
the American Type Culture Collection, Manassas, VA, USA).
Cell culture
H9c2 cardiac myocytes (2×107) were
cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (FBS), 100 µg/ml streptomycin
(Gibco Life Technologies, Carlsbad, CA, USA) and 100 U/ml
penicillin-streptomycin (Gibco Life Technologies) in a humidified
5% CO2 atmosphere at 37°C. H9c2 cardiac myocytes were
passaged every two days and seeded at a density of 2×106
cells/dish in 100-mm dishes with 10% calf serum. The cells were
incubated for 24 h and the medium changed to 0.5% FBS DMEM for a
24-h starvation. To establish whether the protective effects of
H2S were associated with the inhibition of ERK1/2
activity, H9c2 cardiac myocytes were pretreated with 20 µM
U0126 (a selective inhibitor of ERK1/2) for 60 min prior to DOX
treatment.
MTT assay
The MTT assay was used to assess cell viability.
Prior to each experiment, H9c2 cardiac myocytes (5,000 cells/well)
were seeded in 96-well microtiter plates. Following incubation with
U0126 (20 µM) for 60 min and/or NaHS for 30 min, the cells
were treated with 5 µM DOX for a further 24 h. Subsequently,
10 µl MTT solution was added to each well and the microtiter
plates were incubated for 4 h at 37°C. The absorbance was measured
at 470 nm using a SpectraMax 190 spectrophotometer (Molecular
Devices LLC, Sunnyvale, CA, USA) and applied to calculate the
relative ratio of cell viability. Three independent experiments
were performed for each experimental condition.
Assessment of H9c2 cardiac myocyte
apoptosis
Apoptosis was analyzed by fluorescence microscopy
using Hoechst 33258, a chromatin dye. H9c2 cardiac myocytes were
incubated in DMEM containing 0.5% FBS for 24 h (control group), 50
mM DOX for 24 h (DOX group), 100 µM NaHS for 30 min prior to
exposure to 5 µM DOX for 24 h (NaHS + DOX group), 20
µM U0126 for 60 min followed by exposure to 5 µM DOX
for 24 h (U0126 + DOX group), treated with 100 µM NaHS for
30 min followed by a 24-h culture (NaHS group), and treated with 20
µM U0126 for 60 min followed by a 24-h culture (U0126
group). Following various treatments, the cells were fixed in
ice-cold 4% paraformaldehyde (Sigma-Aldrich) dissolved in
phosphate-buffered saline (PBS) at room temperature for 20 min.
Nonspecific binding was blocked using 5% normal goat serum
(Sigma-Aldrich) in 0.01 M PBS containing 0.3% Triton X-100. Cells
were washed twice with PBS and incubated for 15 min with 10
µg/ml Hoechst 33258 at room temperature in the dark. The
cells were visualized under a fluorescence microscope (BX50-FLA;
Olympus Corporation, Tokyo, Japan). Condensed, fractured or
distorted nuclei indicated apoptotic cells, whereas normal nuclear
size and uniform fluorescence were indicative of viable cells.
Measurement of intracellular ROS
levels
The determination of intracellular ROS levels was
performed by measuring a fluorescent product, which was formed by
the oxidation of DCFH-DA. Briefly, the culture medium was removed
and the cells were washed three times with PBS. Following the
addition of fresh culture medium, the cells were incubated at 37°C
for 30 min with DCFH-DA at a final concentration of 10
µmol/l. The cells were washed three further times with PBS
and the relative quantity of fluorescent product was assessed using
the fluorescence microscope connected to an imaging system. The
mean fluorescence intensity (MFI) from five random fields was
measured using ImageJ 1.41o software (National Institutes of
Health, Bethesda, MD, USA) and the MFI served as an index of the
ROS quantity. The experiment was performed in triplicate.
Western blot analysis
The cells were homogenized directly into 10X cell
lysis buffer (Cell Signaling Technology, Inc., Danvers, MA, USA)
and Phosphatase Inhibitor Cocktail (Sigma-Aldrich), lysates were
centrifuged at 12,000 ×g for 10 min at 4°C. The protein
concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology, Haimen, China) according to
the manufacturer's instruction. The extracted proteins were
combined with 5% SDS-PAGE sample buffer (Beyotime Institute of
Biotechnology), boiled at 100°C for 7 min and separated by 10%
SDS-PAGE. Following electrophoresis, the proteins were transferred
to polyvinylidene difluoride membranes. The membranes were blocked
in Tris-buffered saline and Tween-20 (TBS-T; 0.1% Tween-20)
containing 5% non-fat dry milk for 2 h at room temperature with
rotation. Subsequent to blocking, the membranes were incubated with
the following antibodies (all obtained from Cell Signaling
Technology, Inc.) at 4°C overnight: Rabbit anti-ERK1/2 polyclonal
antibody (cat no. 4695P; dilution, 1:2,000), rabbit anti-p-ERK1/2
monoclonal antibody (cat no. 4370P; dilution, 1:2,000) (Cell
Signaling Technology, Inc.), rabbit anti-cystathionine γ-lyase
(CSE) polyclonal antibody (cat no. 12217-1-AP 1:1,000; Proteintech,
Chicago, IL, USA), GAPDH (cat no. AG019; Beyotime Institute of
Biotechnology, Shanghai, China) rabbit anti-Bax polyclonal antibody
(cat no. 2772T; dilution, 1:1,000), and rabbit anti-Bcl-2
polyclonal antibody (cat no. 2870T; dilution, 1:1,000) (Cell
Signaling Technology, Inc.). The membranes were incubated in 5%
milk or bovine serum albumin (Beyotime Institute of Biotechnology)
overnight at 4°C. The membranes were washed three times in TBS-T to
remove the primary antibody, and incubated for 2 h with horseradish
peroxidase-labeled goat anti-rabbit immunoglobulin G (1:1,000,
Beyotime Institute of Biotechnology; cat no. A0208). Following
three washes in TBS-T, the antigen-antibody bands were detected
using an enhanced chemiluminescence reagent kit (Beyotime Institute
of Biotechnology) and quantified using Quantity One densitometry
software, version 4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The western blot data of p-ERK1/2 were presented as a ratio
of the phosphorylated forms to their total forms.
Statistical analysis
Results are presented as means ± standard error of
the mean. Statistical analysis was performed using Student's t-test
or one-way analysis of variance with SPSS 13.0 (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
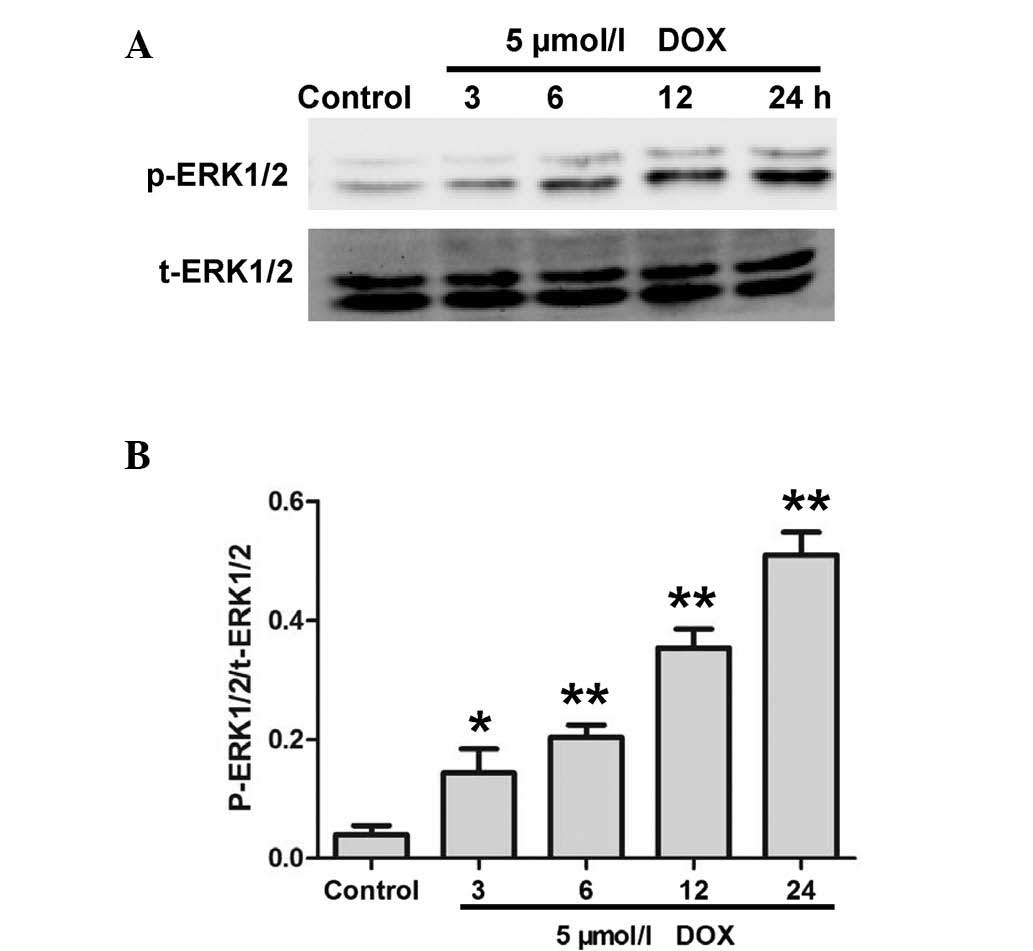
DOX upregulates the expression of
p-ERK1/2 in H9c2 cardiac myocytes
H9c2 cardiac myocytes were treated with DOX for 0,
3, 6, 12 and 24 h to investigate whether DOX treatment exerted an
effect on p-ERK1/2 expression. Western blot analysis revealed that
treatment of H9c2 cardiac myocytes with DOX significantly
upregulated the expression of p-ERK1/2 in a time-dependent manner
(Fig. 1).
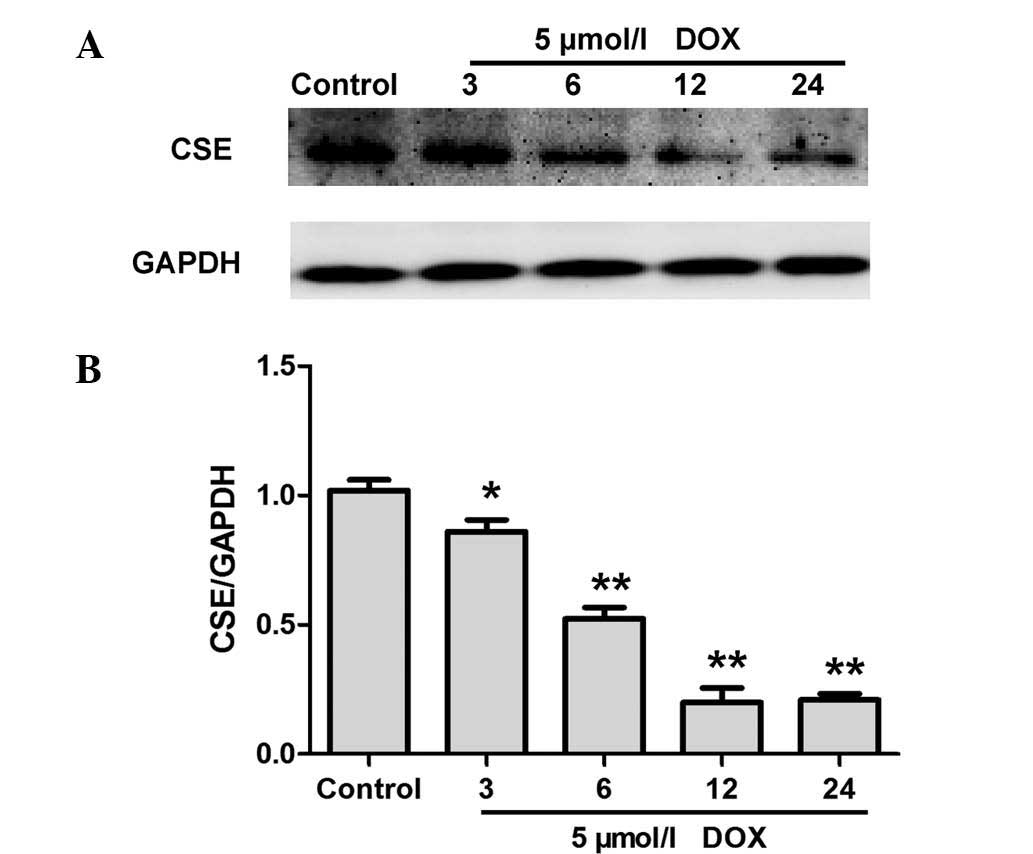
DOX inhibits CSE expression in H9c2
cardiac myocytes
CSE is a major enzyme responsible for endogenous
H2S generation in H9c2 cardiac myocytes (16). Western blot analysis was performed
to evaluate whether DOX decreases endogenous H2S
production by inhibiting the expression of CSE. As shown in
Fig. 2, treatment with DOX for 0,
3, 6, 12 and 24 h resulted in a significant downregulation of CSE
expression in H9c2 cardiac myocytes. These data indicate that DOX
inhibited CSE expression levels in H9c2 cardiac myocytes, and
therefore contributed to a DOX-elicited decrease in endogenous
H2S production.
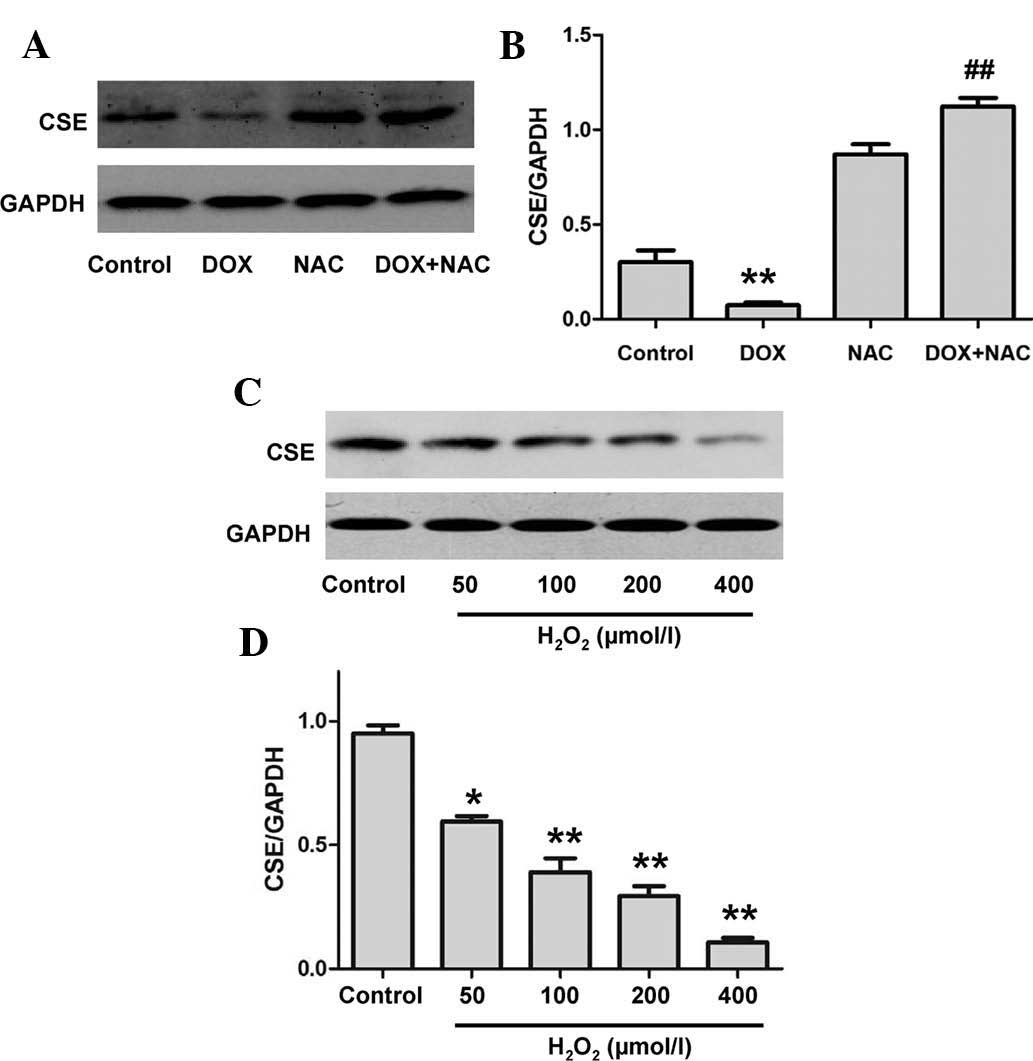
Oxidative stress contributes to
DOX-induced inhibition of CSE expression in H9c2 cardiac
myocytes
Oxidative stress is a primary mechanism by which DOX
induces cardiomyocyte injury. To establish whether oxidative stress
participates in the inhibition of CSE expression caused by DOX
treatment, H9c2 cardiac myocytes were preconditioned with the ROS
scavenger, NAC (1,000 µM) for 60 min prior to DOX treatment.
The results demonstrate that pretreatment of H9c2 cardiac myocytes
with NAC significantly attenuated DOX-induced downregulation of CSE
expression (Fig. 3A and B). In
addition, comparable with the role of DOX, hydrogen peroxide (an
exogenous ROS) was observed to suppress CSE expression (Fig. 3C and D). These data suggest that
oxidative stress contributes to DOX-induced downregulation of CSE
expression in H9c2 cardiac myocytes.
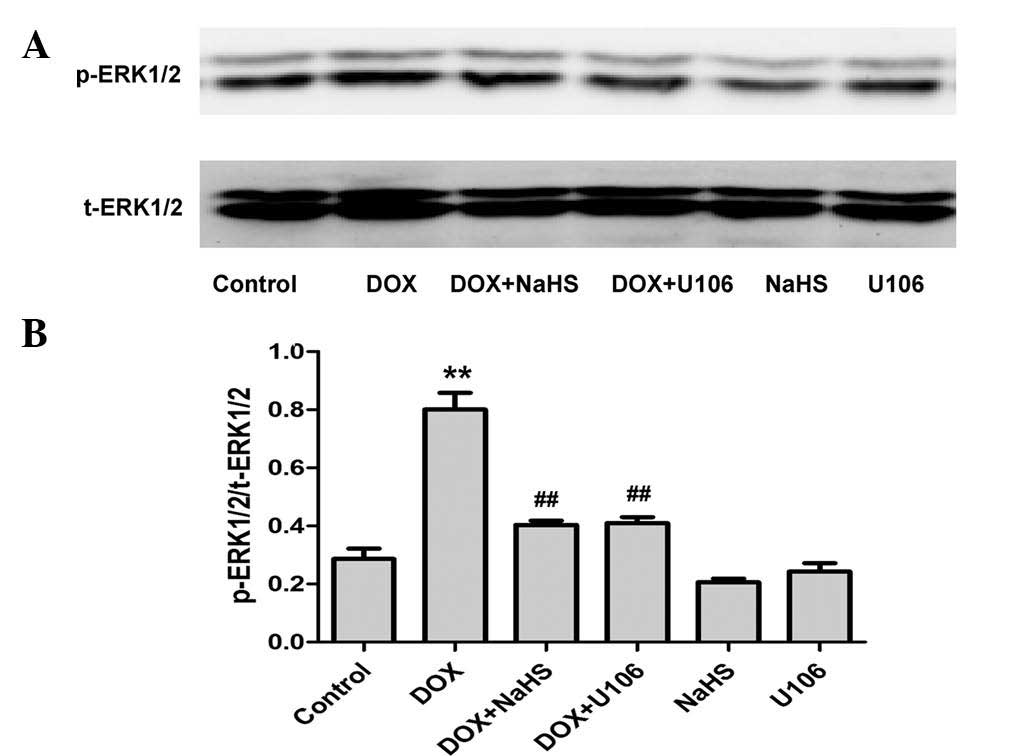
Exogenous H2S inhibits
DOX-induced expression of p-ERK1/2 in H9c2 cardiac myocytes
In order to determine the effect of H2S
on DOX-induced activation of ERK1/2, H9c2 cardiac myocytes were
pretreated with NaHS (a donor of H2S) prior to exposure
to DOX. As demonstrated in Fig. 4,
pretreatment with NaHS significantly attenuated DOX-induced
overexpression of p-ERK1/2. In addition, to further elucidate the
role of ERK1/2 in the cardioprotective action of H2S,
the effect of U0126, a specific ERK1/2 inhibitor, on DOX-induced
expression of p-ERK1/2 was investigated. As shown in Fig. 4, consistent with the effects of
NaHS, pretreatment with 20 µM U0126 for 60 min significantly
attenuated the DOX-induced overexpression of p-ERK1/2. NaHS or
U0126 alone did not exert an effect on the expression of total
(t)-ERK1/2. These data indicate that the cardioprotective action of
H2S is associated with its inhibitory effect on
DOX-induced ERK1/2 activation.
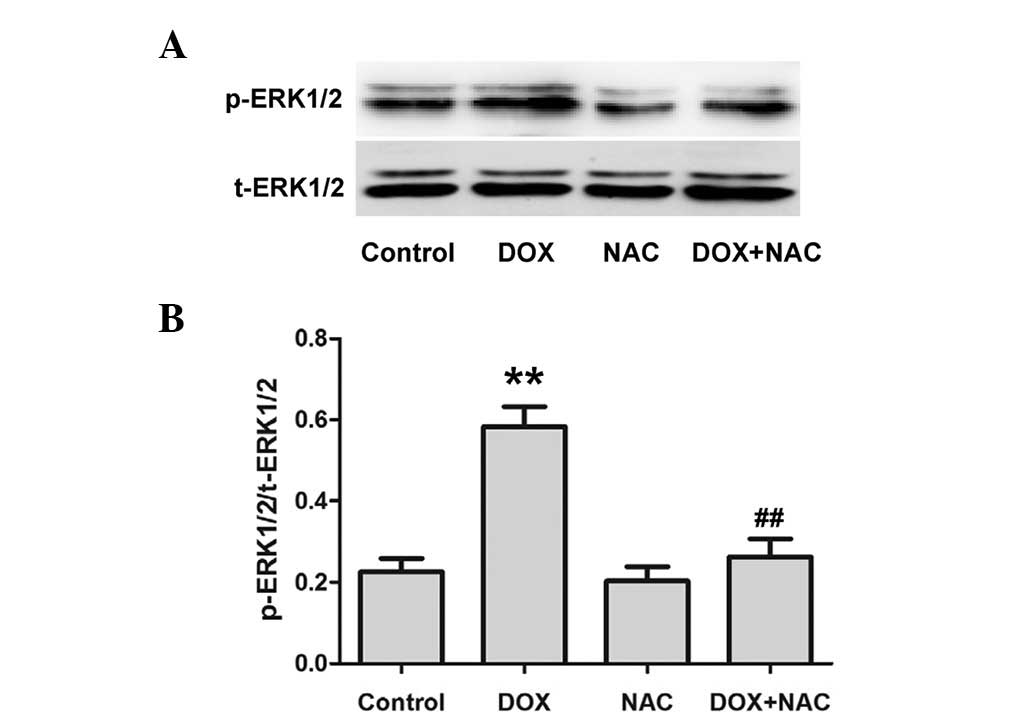
NAC suppresses the DOX-induced expression
of p-ERK1/2 in H9c2 cardiac myocytes
To identify whether the inhibitory effect of NaHS on
the DOX-induced expression of p-ERK1/2 is associated with its
antioxidation, H9c2 cardiac myocytes were pretreated with NAC (a
ROS scavenger) prior to DOX exposure. As shown in Fig. 5, the pretreatment of cells with NAC
significantly attenuated the expression of p-ERK1/2, which is
consistent with the inhibitory effect of NaHS and U0126
pretreatment; however, NAC alone did not significantly alter the
expression levels of t-ERK1/2. These results reveal that an
antioxidant effect may have contributed to the inhibitory effect of
H2S on the DOX-induced expression of p-ERK1/2.
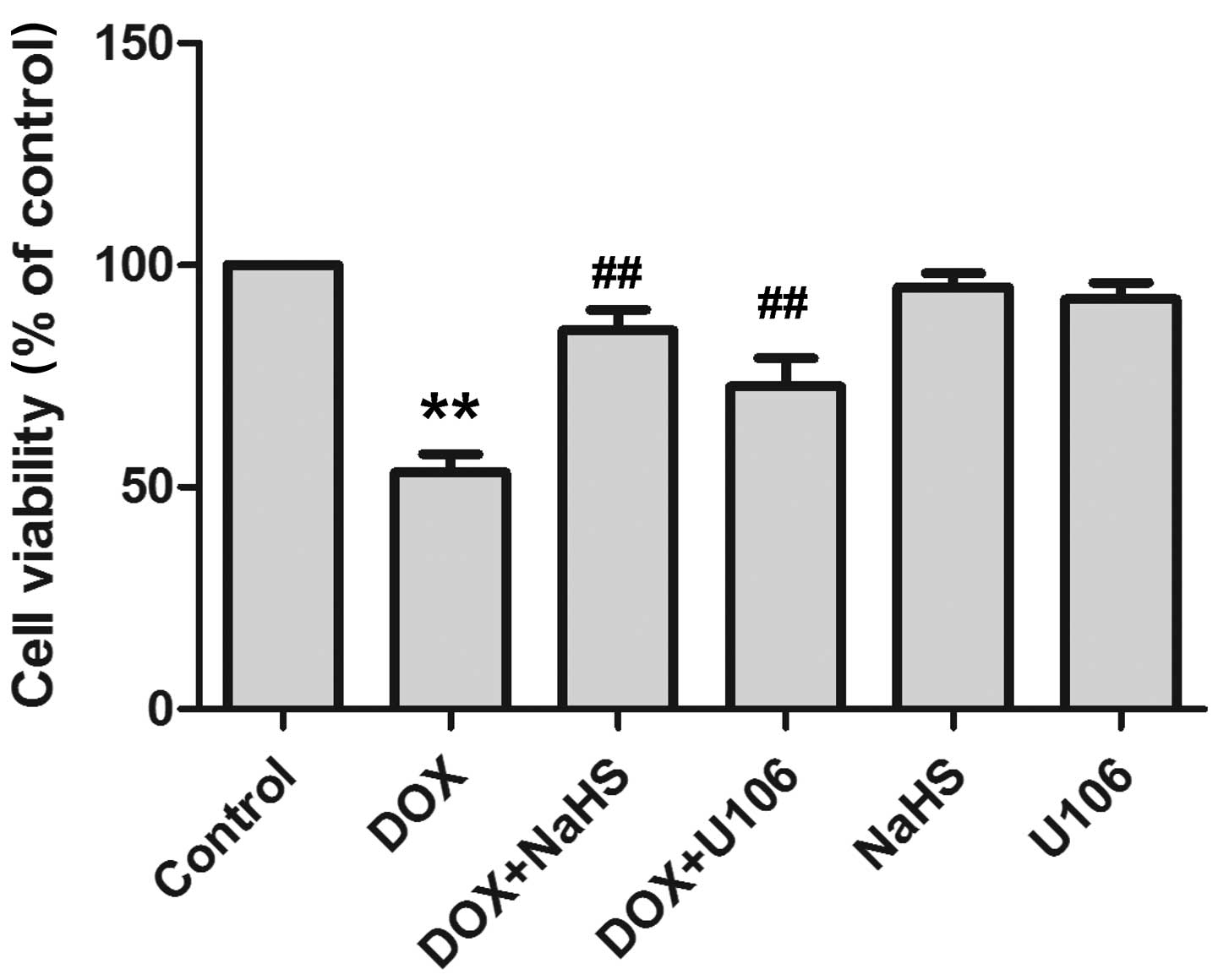
Inhibition of ERK1/2 activation
contributed to protection of H2S against DOX-induced
cytotoxicity
As presented in Fig.
6, exposure of H9c2 cardiac myocytes to DOX resulted in marked
cytotoxicity, leading to a decrease in cell viability. However,
pretreatment of cells with NaHS significantly ameliorated the
DOX-induced cytotoxicity, which was evidenced by an increase in
cell viability. To assess whether the activation of ERK1/2 is
involved in DOX-induced cytotoxicity, H9c2 cardiac myocytes were
pretreated with U0126, a selective inhibitor of ERK1/2. The results
demonstrate that pretreatment with U0126 exerts a similar
cytoprotective effect to H2S against DOX-induced
cytotoxicity. NaHS or U0126 treatment alone was not observed to
alter cell viability in the H9c2 cardiac myocytes. The findings
indicate that H2S blocks DOX-induced cytotoxicity in
H9c2 cardiac myocytes, partially by inhibiting the activation of
ERK1/2.
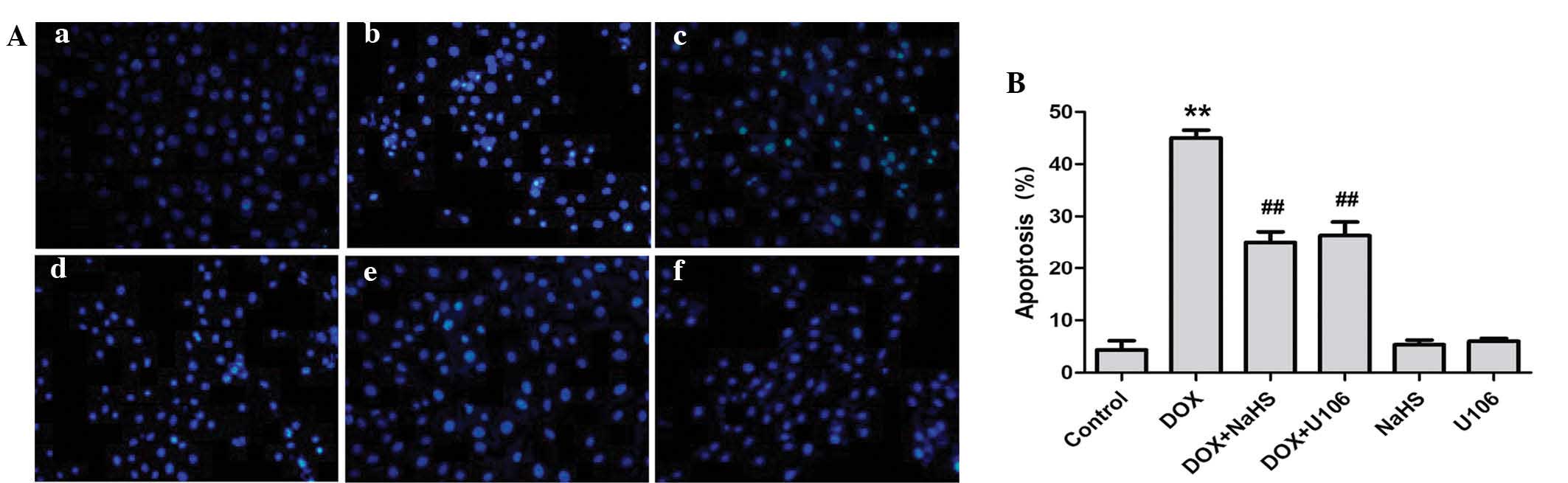
Inhibition of ERK1/2 activation
contributes to the protective effect of H2S against
DOX-induced apoptosis
The effects of NaHS and ERK1/2 inhibition on
DOX-induced apoptosis were investigated further. As shown in
Fig. 7A, H9c2 cardiac myocytes
treated with DOX exhibited typical characteristics of apoptosis,
including condensation of chromatin, shrinkage of nuclei and
apoptotic bodies. However, pretreatment of cells with NaHS markedly
decreased the DOX-induced increased number of cells exhibiting
nuclear condensation and fragmentation. To ascertain whether the
activation of ERK1/2 is implicated in DOX-induced cardiotoxicity,
H9c2 cardiac myocytes were pretreated with U0126. The results
revealed that pretreatment with U0126 attenuated the DOX-induced
increased number of apoptotic H9c2 cardiac myocytes (Fig. 7B). Treatment with NaHS or U0126
alone did not markedly alter H9c2 cell morphology or the percentage
of apoptotic H9c2 cardiac myocytes. These findings demonstrate that
the ERK1/2 signaling pathway participates in DOX-induced
cardiotoxicity.
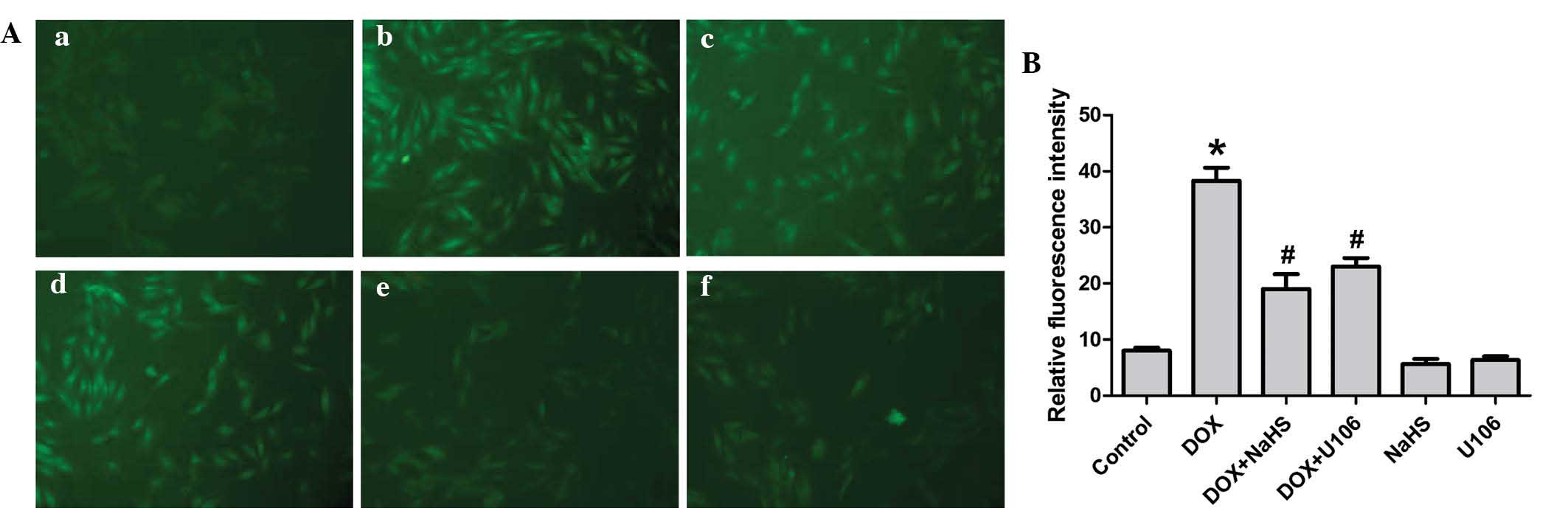
Exogenous H2S and the ERK1/2
inhibitor, U0126, reduce DOX-induced oxidative stress in H9c2
cardiac myocytes
Previous studies have shown that oxidative stress is
critical in DOX-induced cardiotoxicity. Thus, the effects of
H2S and U0126 on DOX-induced ROS generation in H9c2
cardiac myocytes were investigated in the present study. As shown
in Fig. 8, exposure of cells to 5
µM DOX for 24 h significantly enhanced ROS generation.
However, the increased ROS generation was attenuated by
pretreatment with NaHS, indicating that exogenous H2S
protects H9c2 cardiac myocytes against DOX-induced oxidative
stress. To investigate whether the activation of ERK1/2 contributes
to the DOX-induced overproduction of ROS, H9c2 cardiac myocytes
were preconditioned with U0126. The results revealed that
preconditioning with U0126 significantly decreased the DOX-induced
increase in ROS generation. Treatment with NaHS or U0126 alone,
however, did not alter basal ROS generation. The results suggest
that antioxidation of H2S is partly associated with
inhibition of ERK1/2 activation in H9c2 cardiac myocytes.
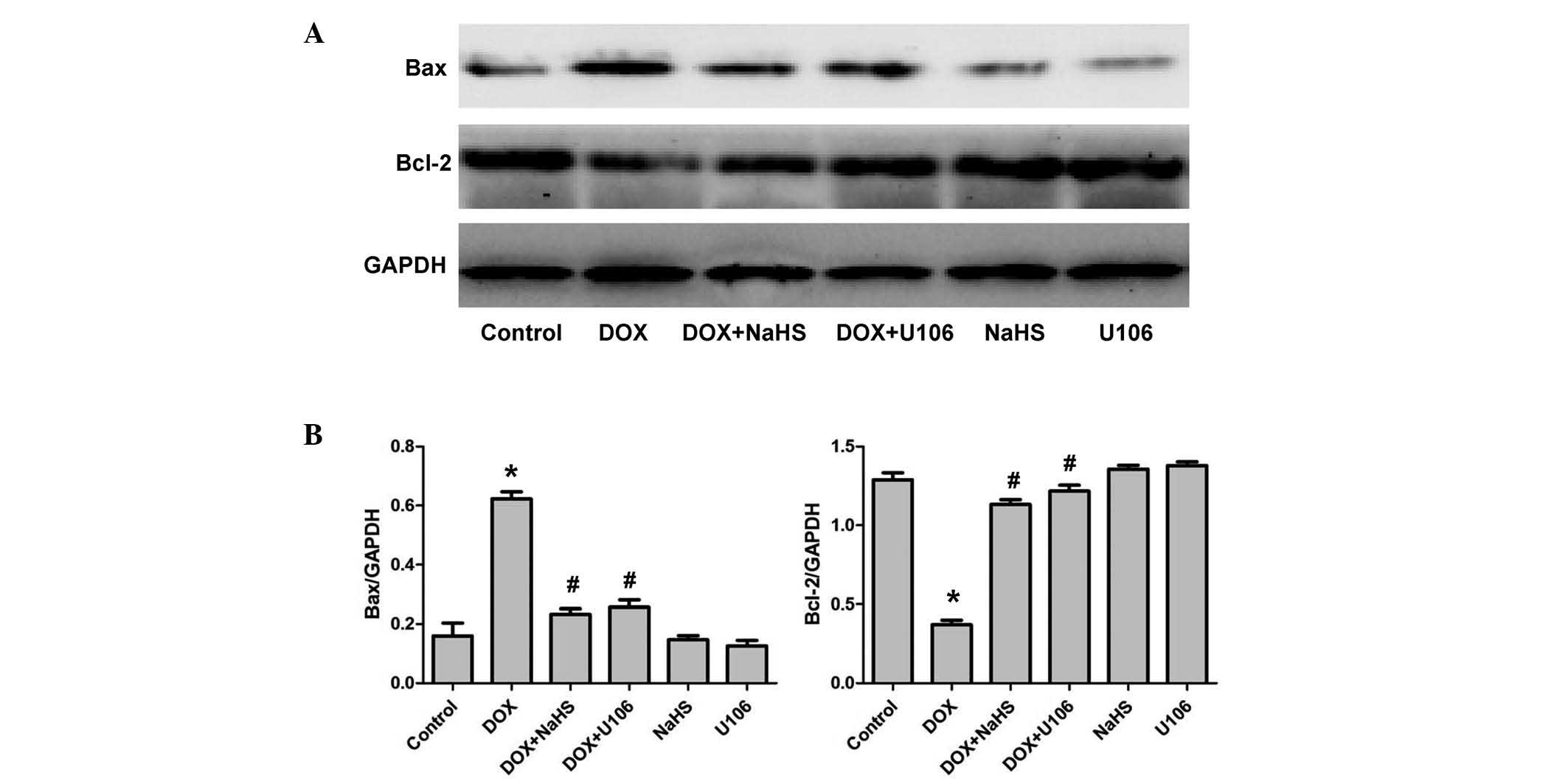
Exogenous H2S and ERK1/2
inhibitor, U0126, inhibit DOX-induced cytotoxicity via upregulation
of Bcl-2 protein expression and downregulation of Bax protein
expression
Bcl-2 is an anti-apoptotic protein and Bax is a
pro-apoptotic protein. To elucidate whether H2S
modulates the expression of Bcl-2 and Bax in DOX-stimulated H9c2
cardiac myocytes, the expression levels of Bcl-2 and Bax protein
were investigated. As presented in Fig. 9, DOX markedly decreased the level
of Bcl-2 expression and increased the level of Bax expression.
However, pretreatment with NaHS or U0126 prior to administration of
DOX, demonstrated that the protein expression levels of Bax were
decreased, whereas the Bcl-2 protein expression levels increased.
These results indicate that H2S prevents apoptosis in
H9c2 cardiac myocytes by upregulating Bcl-2 protein expression and
inhibiting Bax protein expression.
Discussion
Numerous studies have shown that the major molecular
mechanism involved in DOX-induced cardiotoxicity is free
radical-induced oxidative stress and cardiac myocyte death by
apoptosis and necrosis. Concordant with previous studies (17,18),
in the present study, it was observed that exposure of H9c2 cardiac
myocytes to DOX markedly induced cellular injuries, including a
decrease in cell viability, increased cell apoptosis, ROS
generation and activation of ERK1/2.
Previously, the cardioprotective effects of
H2S have been demonstrated in animal models of disease
(19,20). H2S infusion
significantly reduced myocardial infarct size and improved regional
left ventricular function, as well as endothelium-dependent and
-independent micro-vascular reactivity in a porcine model of
myocardial ischemia-reperfusion (I/R) (21). In addition, H2S has been
shown to attenuate myocardial necrosis and apoptosis (22). Endogenous H2S has been
associated with cardioprotection in rat ventricular myocytes as a
result of metabolic inhibition preconditioning (23). Furthermore, inhibition of
endogenous H2S generation by inhibition of its synthesis
inhibitor has been shown to block the protective effect of IPC in
isolated hearts, as well as isolated cardiac myocytes (24). In the present study, H9c2 cardiac
myocytes were used to investigate the effect of DOX on endogenous
H2S generation and its role in the cardiotoxicity of
DOX. Exposure of H9c2 cardiac myocytes to DOX was observed to
result in a significant decrease in H2S generation.
ERK1/2 is important in cell proliferation, growth
and cell death (25). Previous
research indicates that the ERK1/2 signaling pathway is activated
by DOX-induced apoptosis in H9c2 cardiac myocytes (26). Furthermore, ERK activation has been
demonstrated to be important in certain models that induce
apoptosis in the myocardium, including isoproterenol-induced
apoptosis (27) and I/R in
neonatal cardiomyocytes that induce apoptosis (28). In the current study, the results
showed that the expression level of p-ERK1/2 was increased
following DOX-induced injury in H9c2 cardiac myocytes, and
H2S treatment decreased the expression level of
p-ERK1/2, and subsequently inhibited DOX-induced injuries in H9c2
cardiac myocytes. Additionally, the present study further
demonstrated that the ERK1/2 inhibitor, U0126 markedly reduced
DOX-induced injuries (which was evidenced by an increase in cell
viability), decreased the expression level of p-ERK1/2, and
attenuated DOX-induced apoptosis in H9c2 cardiac myocytes. These
results indicate that inhibition of the ERK1/2 signaling pathway
may be involved in the protection of exogenous H2S.
Notably, the present study further demonstrated that
ROS were involved in DOX-induced cell injuries and whether DOX
activation of ERK1/2 is due to its induction of ROS was
investigated. It was shown that pretreatment of H9c2 cardiac
myocytes with NAC (a ROS scavenger) significantly attenuated
DOX-induced expression of p-ERK1/2. Collectively, the results of
the present study support the hypothesis that DOX induction of ROS
activates ERK1/2, which mediates DOX-induced injuries in H9c2
cardiac myocytes.
The effect of H2S on regulating the
intracellular Bcl-2/Bax signaling pathway was investigated in the
present study to provide further biochemical evidence elucidating
the protective effect of H2S on cardiac myocytes against
DOX-induced injuries. Bcl-2 is an oncogene-derived protein, which
confers negative control in the signaling pathway of cellular
suicide machinery (29). Bax is a
Bcl-2 homologous protein, which promotes cell death by competing
with Bcl-2 (30). Compared with
the controls, DOX exposure was observed to downregulate the protein
expression of Bcl-2, while it increased Bax protein expression in
H9c2 cardiac myocytes. Furthermore, H2S supplementation
in H9c2 cardiac myocytes significantly reduced DOX-induced Bax
expression and augmented DOX-suppressed Bcl-2 expression, and these
effects were associated with a decrease in apoptotic levels. The
results suggested a potent protective effect by H2S
against DOX-induced injuries, partly through upregulation of Bcl-2
and downregulation of Bax.
In conclusion, the principal finding of the current
study was that H2S inhibits DOX-induced cardiotoxicity
in H9c2 cardiac myocytes, and its effects may involve inhibition of
ROS-mediated activation of ERK1/2, upregulation of Bcl-2 and
downregulation of Bax. The present study elucidated the underlying
mechanisms of H2S protection against DOX-induced
cardiotoxicity, and provided valuable evidence for identifying
H2S as a novel therapeutic strategy for the treatment
and prevention of DOX-induced cardiomyopathy.
Acknowledgments
The present study was supported by grants provided
by the Medical Scientific Research Fund of Guangdong Province
(grant no. A2014810) and the Graduate Student Research Innovation
Project of Hunan Province (grant no. CX2013B397).
References
|
1
|
Menna P, Recalcati S, Cairo G and Minotti
G: An introduction to the metabolic determinants of anthracycline
cardiotoxicity. Cardiovasc Toxicol. 7:80–85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lipshultz SE, Karnik R, Sambatakos P,
Franco VI, Ross SW and Miller TL: Anthracycline-related
cardiotoxicity in childhood cancer survivors. Curr Opin Cardiol.
29:103–112. 2014. View Article : Google Scholar
|
|
3
|
Spallarossa P, Garibaldi S, Altieri P,
Fabbi P, Manca V, Nasti S, Rossettin P, Ghigliotti G, Ballestrero
A, Patrone F, et al: Carvedilol prevents doxorubicin-induced free
radical release and apoptosis in cardiomyocytes in vitro. J Mol
Cell Cardiol. 37:837–846. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu MH, Zhang Y, Lin XL, He J, Tan TP, Wu
SJ, Yu S, Chen L, Chen YD, Fu HY, et al: Hydrogen sulfide
attenuates doxorubicin-induced cardiotoxicity through inhibiting
calreticulin expression in H9c2 cells. Mol Med Rep. Jul 2–2015.Epub
ahead of print. View Article : Google Scholar
|
|
5
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang YE, Tang ZH, Xie W, Shen XT, Liu MH,
Peng XP, Zhao ZZ, Nie DB, Liu LS and Jiang ZS: Endogenous hydrogen
sulfide mediates the cardioprotection induced by ischemic
post-conditioning in the early reperfusion phase. Exp Ther Med.
4:1117–1123. 2012.PubMed/NCBI
|
|
7
|
Grisanti LA, Talarico JA, Carter RL, Yu
JE, Repas AA, Radcliffe SW, Tang HA, Makarewich CA, Houser SR and
Tilley DG: β-Adrenergic receptor-mediated transactivation of
epidermal growth factor receptor decreases cardiomyocyte apoptosis
through differential subcellular activation of ERK1/2 and Akt. J
Mol Cell Cardiol. 72:39–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu W, Wu W, Chen J, Guo R, Lin J, Liao X
and Feng J: Exogenous hydrogen sulfide protects H9c2 cardiac cells
against high glucose-induced injury by inhibiting the activities of
the p38 MAPK and ERK1/2 pathways. Int J Mol Med. 32:917–925.
2013.PubMed/NCBI
|
|
9
|
Lou H, Danelisen I and Singal PK:
Involvement of mitogen-activated protein kinases in
adriamycin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol.
288:H1925–H1930. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu J, Mao W, Ding B and Liang CS:
ERKs/p53 signal transduction pathway is involved in doxorubicin
induced apoptosis in H9c2 cells and cardiomyocytes. Am J Physiol
Heart Circ Physiol. 295:H1956–H1965. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Du J, Hui Y, Cheung Y, Bin G, Jiang H,
Chen X and Tang C: The possible role of hydrogen sulfide as a
smooth muscle cell proliferation inhibitor in rat cultured cells.
Heart Vessels. 19:75–80. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, Kim
HR, Jeon SB, Jeon WK, Chae HJ and Chung HT: Hydrogen sulfide
inhibits nitric oxide production and nuclear factor-kappaB via heme
oxygenase-1 expression in RAW264.7 macrophages stimulated with
lipopolysaccharide. Free Radic Biol Med. 41:106–119. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Adhikari S and Bhatia M:
H2S-induced pancreatic acinar cell apoptosis is mediated
via JNK and p38 MAP kinase. J Cell Mol Med. 12:1374–1383. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang G, Yang W, Wu L and Wang R:
H2S, endoplasmic reticulum stress and apoptosis of
insulin-secreting beta cells. J Biol Chem. 282:16567–16576. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo R, Lin J, Xu W, Shen N, Mo L, Zhang C
and Feng J: Hydrogen sulfide attenuates doxorubicin-induced
cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells.
Int J Mol Med. 31:644–650. 2013.PubMed/NCBI
|
|
16
|
Kimura H: Hydrogen sulfide: Its
production, release and functions. Amino Acids. 41:113–121. 2011.
View Article : Google Scholar
|
|
17
|
Wang X, Wang XL, Chen HL, Wu D, Chen JX,
Wang XX, Li RL, He JH, Mo L, Cen X, et al: Ghrelin inhibits
doxorubicin cardio-toxicity by inhibiting excessive autophagy
through AMPK and p38-MAPK. Biochem Pharmacol. 88:334–350. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo R, Wu K, Chen J, Mo L, Hua X, Zheng D,
Chen P, Chen G, Xu W and Feng J: Exogenous hydrogen sulfide
protects against doxorubicin-induced inflammation and cytotoxicity
by inhibiting p38MAPK/NFkappaB pathway in H9c2 cardiac cells. Cell
Physiol Biochem. 32:1668–1680. 2013.
|
|
19
|
Łowicka E and Bełtowski J: Hydrogen
sulfide (H2S)-the third gas of interest for
pharmacologists. Pharmacol Rep. 59:4–24. 2007.
|
|
20
|
Ji Y, Pang QF, Xu G, Wang L, Wang JK and
Zeng YM: Exogenous hydrogen sulfide postconditioning protects
isolated rat hearts against ischemia-reperfusion injury. Eur J
Pharmacol. 587:1–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Osipov RM, Robich MP, Feng J, Liu Y,
Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C and Sellke FW:
Effect of hydrogen sulfide in a porcine model of myocardial
ischemia-reperfusion: comparison of different administration
regimens and characterization of the cellular mechanisms of
protection. J Cardiovasc Pharmacol. 54:287–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sodha NR, Clements RT, Feng J, Liu Y,
Bianchi C, Horvath EM, Szabo C and Sellke FW: The effects of
therapeutic sulfide on myocardial apoptosis in response to
ischemia-reperfusion injury. Eur J Cardiothorac Surg. 33:906–913.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan TT, Feng ZN, Lee SW, Moore PK and Bian
JS: Endogenous hydrogen sulfide contributes to the cardioprotection
by metabolic inhibition preconditioning in the rat ventricular
myocytes. J Mol Cell Cardiol. 40:119–130. 2006. View Article : Google Scholar
|
|
24
|
Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY,
Zhou S and Moore PK: Role of hydrogen sulfide in the
cardioprotection caused by ischemic preconditioning in the rat
heart and cardiac myocytes. J Pharmacol Exp Ther. 316:670–678.
2006. View Article : Google Scholar
|
|
25
|
Javadov S, Jang S and Agostini B:
Crosstalk between mitogenactivated protein kinases and mitochondria
in cardiac diseases: Therapeutic perspectives. Pharmacol Ther.
144:202–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Mao W, Ding B and Liang CS:
ERKs/p53 signal transduction pathway is involved in
doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am
J Physiol Heart Circ Physiol. 295:H1956–H1965. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou B, Wu LJ, Tashiro S, Onodera S,
Uchiumi F and Ikejima T: Activation of extracellular
signal-regulated kinase during silibinin-protected,
isoproterenol-induced apoptosis in rat cardiac myocytes is tyrosine
kinase pathway-mediated and protein kinase C-dependent. Acta
Pharmacol Sin. 28:803–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang CM, Han LP, Li HZ, Qu YB, Zhang ZR,
Wang R, Xu CQ and Li WM: Calcium-sensing receptors induce apoptosis
in cultured neonatal rat ventricular cardiomyocytes during
simulated ischemia/reperfusion. Cell Biol Int. 32:792–800. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gómez-Fernández JC: Functions of the
C-terminal domains of apoptosis-related proteins of the Bcl-2
family. Chem Phys Lipids. 183:77–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Renault TT and Manon S: Bax: Addressed to
kill. Biochimie. 93:1379–1391. 2011. View Article : Google Scholar : PubMed/NCBI
|