Introduction
Ventilator-associated pneumonia (VAP), the most
common type of hospital-acquired pneumonia (HAP), is defined as
bacterial pneumonia that develops in patients who have received
mechanical ventilation for >48 h (1). VAP occurs in 6–52% of mechanically
ventilated patients and in certain specific settings, the incidence
is up to 76% (2). The incidence of
VAP varies depending on the type of population studied, the
intensity of preventive measures implemented and the presence or
absence of risk factors. With an estimated attributable mortality
of 9%, VAP usually increases the time of mechanical ventilation and
time spent in the intensive care unit (ICU) by >10 days and its
treatment accounts for at least 50% of antibiotics prescribed in
ICUs, producing a huge cost (3).
The expected burden of VAP in the coming years may increase with
the increasing age of the population, intensification of care and
the growing prevalence of severe underlying diseases in ICU
patients (4). As the diagnosis as
well as the treatment of VAP remain challenging, studies are
required to gain a comprehensive understanding of VAP.
A number of previous studies have shown that up- or
down-regulation of the expression of certain genes is closely
associated with the development of infection. For instance, a study
on the polymorphism of tumor necrosis factor (TNF)-α reported that
overexpression of TNF-α is associated with a 2.1-13-fold increase
in the incidence of severe sepsis from all causes, including
pneumonia (5). PI3, which
encodes an elastase-specific inhibitor with antimicrobial peptide
activity, has proven to promote early clearance of Pseudomonas
aeruginosa by activating macrophages and recruiting neutrophils
(6). PIK3R3, which encodes
the protein of phosphoinositide 3-kinase regulatory sub-unit gamma,
was shown to be predominately expressed in immune cells and to be
involved in chemoattractant-induced cell migration (7). None of these genes alone, however, is
sufficient enough to answer the fundamental question why certain
patients develop VAP, while other similar patients do not.
As a genome-wide screening approach, microarray
analysis may be effective for identifying novel genetic factors or
gene expression profiles associated with the development of
infection in VAP. With the goal to construct a model for the
prediction of susceptibility to VAP in critically-injured trauma
patients, Swanson et al (8)
analyzed the gene expression profiles of patients who developed VAP
and those who were never affected. By comparing the differentially
expressed genes (DEGs) between the two groups, a logistic
regression model was constructed, which comprised five genes
(PIK3R3, ATP2A1, PI3, ADAM8 and HCN4) and was able to
accurately categorize 95% of patients that developed VAP.
The present study identified DEGs between patients
with VAP and those without VAP based on the expression profiling
data provided by Swanson et al (8) and then performed functional
annotation and enrichment analyses in order to elucidate the
potential molecular mechanisms of VAP. In addition, a
protein-protein interaction (PPI) network was constructed and the
functional modules in this network were analyzed. The novel view on
VAP provided by the present study indicates that VAP requires
further study.
Materials and methods
Derivation of genetic data
The gene expression profile dataset GSE30385
deposited by Swanson et al (8) was downloaded from the Gene Expression
Omnibus database (http://www.ncbi.nlm.nih.gov/geo), a public functional
genomics data repository. The annotation platform was the GPL201
(HG-Focus) Affymetrix Human HG-Focus Target Array platform
(Affymetrix, Inc., Santa Clara, CA, USA) updated on May 10th, 2013.
A total of 20 samples from critically ill trauma patients were
available, including 10 patients who developed
ventilator-associated pneumonia (VAP group) and 10 who did not
(NoVAP group).
Data processing
The Robust Multichip Average method of the
R/Bioconductor Affy package (http://www.bioconductor.org/; Affymetrix, Inc.) was
used to pre-process the downloaded raw data via background
adjustment, quintile normalization and summarization (9). Using the R/Bioconductor software v
3.1 (http://www.bioconductor.org/), probe
serial numbers in the matrix were transformed into gene names. As
certain different probes can be mapped to the same gene, the
average probe expression value was calculated as the final
expression value of the corresponding gene.
Screening of DEGs
To screen DEGs between the VAP group and the NoVAP
group, the Linear Models for Microarray Data (Limma) package in
R/Bioconductor software 3.1 (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
was used (10). The
Benjamini-Hochberg false discovery rate (FDR) was used to adjust
the raw P-value in the multiple testing (11,12).
DEGs were screened with cutoff values of FDR<0.05 and |log (fold
change)|≥1.5 (13,14). The eligible DEGs were grouped into
upregulated and downregulated genes. To guarantee that the
identified DEGs represent the two types of specimen, cluster
analysis of DEGs was also performed using the Pheatmap package of R
(http://cran.r-project.org/web/packages/pheatmap/index.html).
Functional annotation and pathway
enrichment analysis
The Database for Annotation Visualization and
Integrated Discovery (DAVID) provides a comprehensive set of
functional annotation tools to understand the biological meaning of
large lists of genes by using a hypergeometric distribution
algorithm (15). To functionally
annotate DEGs identified between the VAP group and the NoVAP group,
DAVID was used to analyze the significantly enriched Gene Ontology
(GO) terms (16) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways (17) for upregulated and down-regulated
genes, respectively (adjusted P-value <0.05).
Computational identification of
transcription factors (TF), tumor suppressor genes (TSGs) and
oncogenes
TRANSFAC version 7.0 (http://www.gene-regulation.com/pub/databases.html),
which was initially a data collection, has evolved into the basis
for a complex platform for the description and analysis of gene
regulatory events and networks, including TFs as well as their
DNA-binding sites and profiles (18). The TSGene database was designed by
integrating TSGs with large-scale experimental evidence to offer a
comprehensive resource for further investigation of TSGs and their
molecular mechanisms in cancer (19). The tumor-associated gene (TAG)
database was developed by integrating information from
well-characterized oncogenes and tumor suppressor genes to
facilitate cancer research (20).
Based on these three databases, DEGs identified between the VAP
group and the NoVAP group were analyzed for the screening of TFs,
TSGs and TAGs.
Protein-protein interaction (PPI) network
construction
The Search Tool for the Retrieval of Interacting
Genes (STRING version 1.0; http://string-db.org/) is an online database which
includes experimental as well as predicted interaction information
and comprises >1,100 completely sequenced organisms (21). The DEGs identified above were
directly mapped to the STRING database for acquiring significant
PPI pairs from a range of sources, including data from experimental
studies and data retrieved by text mining and homology searches.
PPI pairs with the combined score of ≥0.4 were retained for the
construction of the PPI network.
Functional module analysis
CFinder (http://www.cfinder.org/) is a fast program for
locating and visualizing overlapping, densely interconnected groups
of nodes (defined as modules) in undirected graphs, and allows for
easy navigation between the original graph and the network of these
groups (22). Using the default
parameter of k=3, modules were screened from the PPI network
constructed as described above using CFinder. The top two
sub-modules identified by modularity analysis were then selected
for later functional enrichment by DAVID (FDR<0.05).
Results
Identification of DEGs
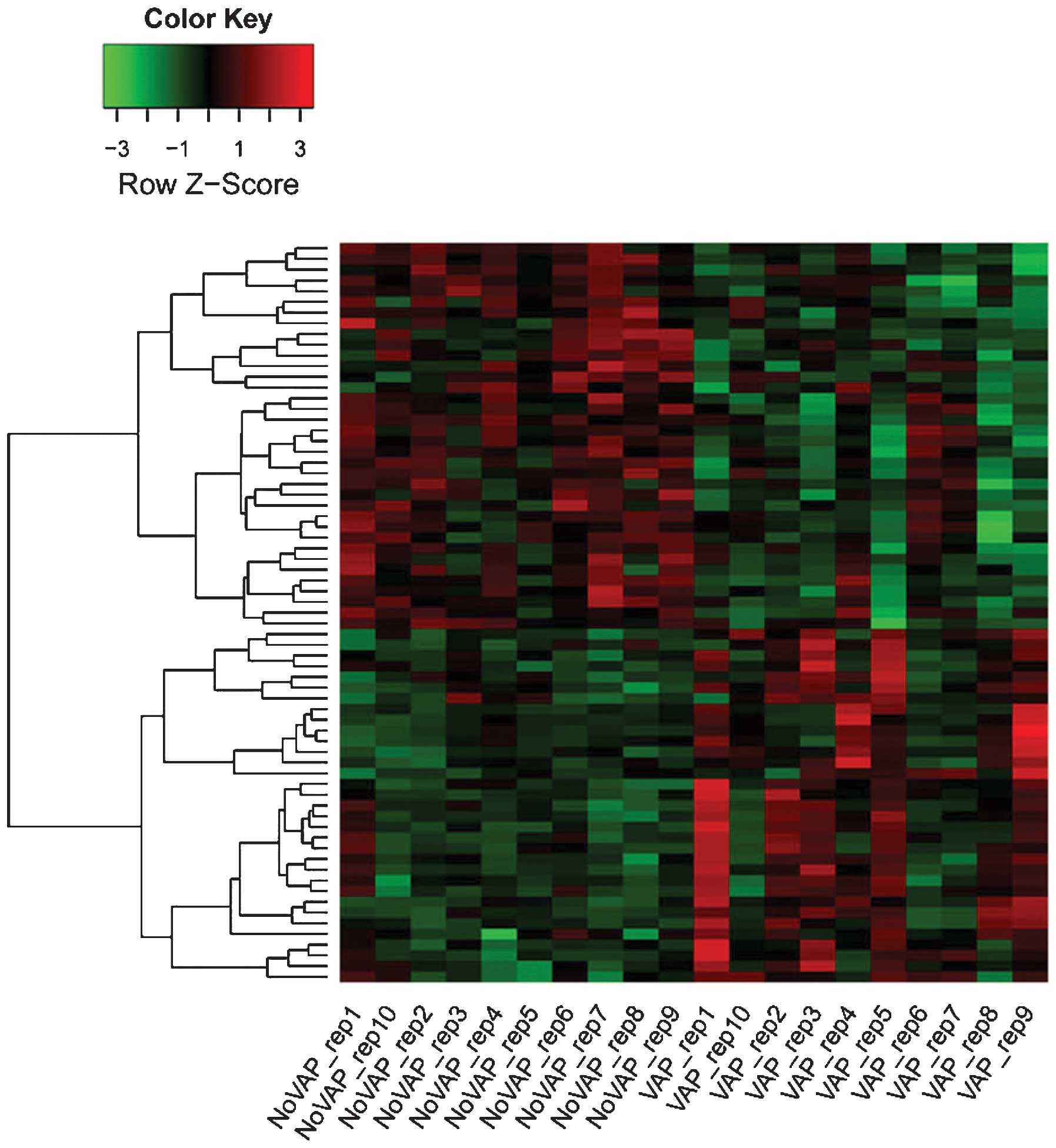
With a FDR<0.05 and |log (fold change)|≥1.5, 69
DEGs between the VAP group and the NoVAP group were obtained. Of
these DEGs, 33 (47.83%) were upregulated and 36 (52.17%) were
downregulated in the VAP group compared with the NoVAP group. The
results of the clustering analysis of the DEGs are shown in
Fig. 1.
Functional annotation and pathway
enrichment analysis
To identify the functions of these DEGs, all of the
up- and downregulated genes were mapped to terms of the GO
database, which consists of three ontologies, namely biological
processes (BP), cellular components (CC) and molecular function
(MF). In the three categories of the GO terms, 'immune system
processes', 'immune response', 'kinase activity' were dominant for
the upregulated genes, while 'response to stress', 'programmed cell
death' and 'peptidase inhibitor activity' were dominant among the
downregulated genes (Table I).
 | Table IClassification of differentially
expressed genes in ventilator-associated pneumonia according to GO
terms with P<0.05. |
Table I
Classification of differentially
expressed genes in ventilator-associated pneumonia according to GO
terms with P<0.05.
| Gene category | GO ID | Function | Count | P-value |
|---|
| Upregulated
genes | | | | |
| BP | GO:0009617 | Response to
bacterium | 9 |
6.66×10−8 |
| GO:0006955 | Immune
response | 14 |
2.12×10−7 |
| GO:0051707 | Response to other
organism | 9 |
5.28×10−6 |
| GO:0009607 | Response to biotic
stimulus | 9 |
7.59×10−6 |
| GO:0002376 | Immune system
process | 15 |
9.69×10−6 |
| CC | GO:0005576 | Extracellular
region | 13 | 0.0001 |
| GO:0071682 | Endocytic vesicle
lumen | 2 | 0.0004 |
| GO:0030139 | Endocytic
vesicle | 4 | 0.0005 |
| GO:0005615 | Extracellular
space | 6 | 0.0052 |
| GO:0045335 | Phagocytic
vesicle | 2 | 0.0076 |
| MF | GO:0004908 | Interleukin-1
receptor activity | 2 |
8.24×10−5 |
| GO:0035251 |
UDP-glucosyltransferase activity | 2 | 0.0001 |
| GO:0046527 | Glucosyltransferase
activity | 2 | 0.0002 |
| GO:0004708 | Kinase
activity | 2 | 0.0005 |
| GO:0004712 | Protein
serine/threonine/tyrosine kinase activity | 2 | 0.0023 |
| Downregulated
genes | | | | |
| BP | GO:0050873 | Brown fat cell
differentiation | 3 |
5.08×10−5 |
| GO:0006950 | Response to
stress | 18 | 0.0001 |
| GO:0050790 | Regulation of
catalytic activity | 12 | 0.0001 |
| GO:0006915 | Apoptotic
process | 12 | 0.0002 |
| GO:0012501 | Programmed cell
death | 12 | 0.0002 |
| CC | GO:0044445 | Cytosolic part | 4 | 0.0005 |
| GO:0022626 | Cytosolic
ribosome | 3 | 0.0009 |
| GO:0044421 | Extracellular
region part | 8 | 0.0019 |
| GO:0044391 | Ribosomal
subunit | 3 | 0.0026 |
| GO:0005829 | Cytosol | 12 | 0.0036 |
| MF | GO:0042277 | Peptide
binding | 4 | 0.0004 |
| GO:0033218 | Amide binding | 4 | 0.0005 |
| GO:0004866 | Endopeptidase
inhibitor activity | 4 | 0.0006 |
| GO:0061135 | Endopeptidase
regulator activity | 4 | 0.0006 |
| GO:0030414 | Peptidase inhibitor
activity | 4 | 0.0006 |
KEGG was used to further identify the altered
biological functions arising from these DEGs. The upregulated genes
were mainly enriched in ten pathways, including the neurotrophin
signaling pathway, the mitogen-activated protein kinase (MAPK)
signaling pathway and the nucleotide-binding oligomerization domain
(NOD)-like receptor signaling pathway, while the downregulated
genes were enriched in nine pathways, which included complement and
coagulation cascades as well as pathways in cancer and ribosomes
(Table II).
| Table IIEnrichment of KEGG pathways in
ventilator-associated pneumonia with P<0.05. |
Table II
Enrichment of KEGG pathways in
ventilator-associated pneumonia with P<0.05.
| Gene category | KEGG ID | Function | Count | P-value |
|---|
| Upregulated
genes | 4722 | Neurotrophin
signaling pathway | 3 | 0.0064 |
| 4010 | Mitogen-activated
protein kinase signaling | 4 | 0.0078 |
| 5014 | Amyotrophic lateral
sclerosis | 2 | 0.0112 |
| 4621 | Nucleotide-binding
oligomerization domain | 2 | 0.0133 |
| | receptor signaling
pathway | | |
| 4664 | Fc epsilon RI
signaling pathway | 2 | 0.0238 |
| 4640 | Hematopoietic cell
lineage | 2 | 0.0291 |
| 4912 |
Gonadotropin-releasing hormone
signaling | 2 | 0.0375 |
| 4620 | Toll-like receptor
signaling pathway | 2 | 0.0382 |
| 5146 | Amoebiasis | 2 | 0.0410 |
| 4060 | Cytokine-cytokine
receptor interaction | 3 | 0.0449 |
| Downregulated
genes | 4610 | Complement and
coagulation cascades | 4 | 0.0001 |
| 3010 | Ribosome | 3 | 0.0053 |
| 5150 | Staphylococcus
aureus infection | 2 | 0.0192 |
| 5131 | Shigellosis | 2 | 0.0234 |
| 5140 | Leishmaniasis | 2 | 0.0318 |
| 5220 | Chronic myeloid
leukemia | 2 | 0.0326 |
| 5200 | Pathways in
cancer | 4 | 0.0358 |
| 5222 | Small cell lung
cancer | 2 | 0.0431 |
| 4012 | ErbB signaling
pathway | 2 | 0.0449 |
Identification of TFs, TSGs and
oncogenes
According to the TRANSFAC, TSGene and TAG databases,
a total of four TFs, two oncogenes, four TSGs and two other TAGs
were further screened from the DEGs preliminarily identified
between the VAP group and the NoVAP group (Table III).
| Table IIIAnnotation for TFs, TSGs, oncogenes
and other TAGs in ventilator-associated pneumonia. |
Table III
Annotation for TFs, TSGs, oncogenes
and other TAGs in ventilator-associated pneumonia.
| Gene category | TFs | Oncogenes | TSGs | Other TAGs |
|---|
| Upregulated
genes | KLF7 | CD24 | LTF | NA |
| Downregulated
genes | EGR3, FOSB,
NCOR2 | CRK | BTG2, CDKN1A,
THBD | RGS2, CTSZ |
PPI construction
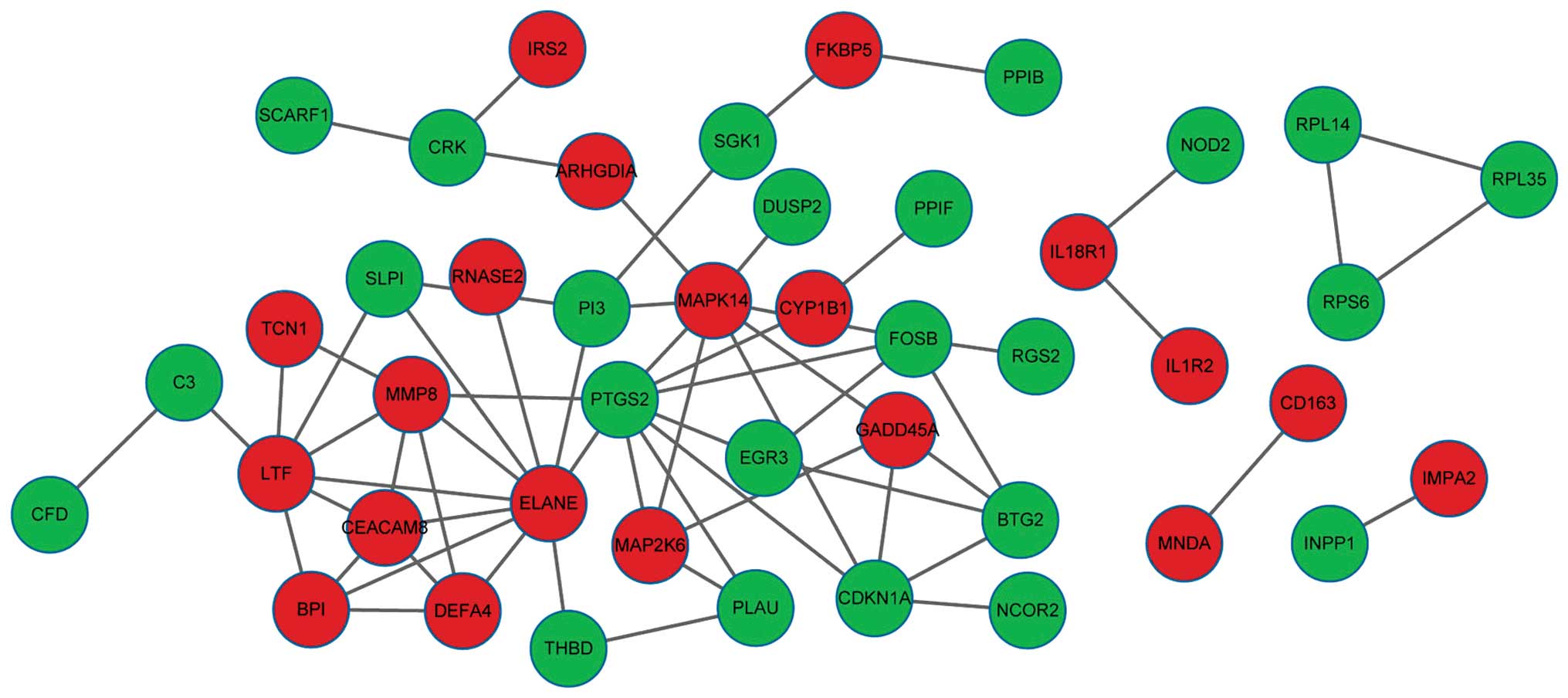
The PPI network contained 64 PPI pairs and 44 nodes
(20 upregulated genes and 24 downregulated genes in the VAP group)
(Fig. 2). The five node proteins
with a connection degree of >5 were ELANE (connection degree,
10), PTGS2 (connection degree, 9), MAPK14 (connection degree, 8),
LTF (connection degree, 7) and MMP8 (connection degree, 6).
Functional module analysis
High aggregation, which reflects the high
modularization of a gene network, is an important characteristic of
biological networks. To distinguish the modules with specific
functions and different sizes, the constructed network was usually
divided into relatively independent sub-modules prior to analysis.
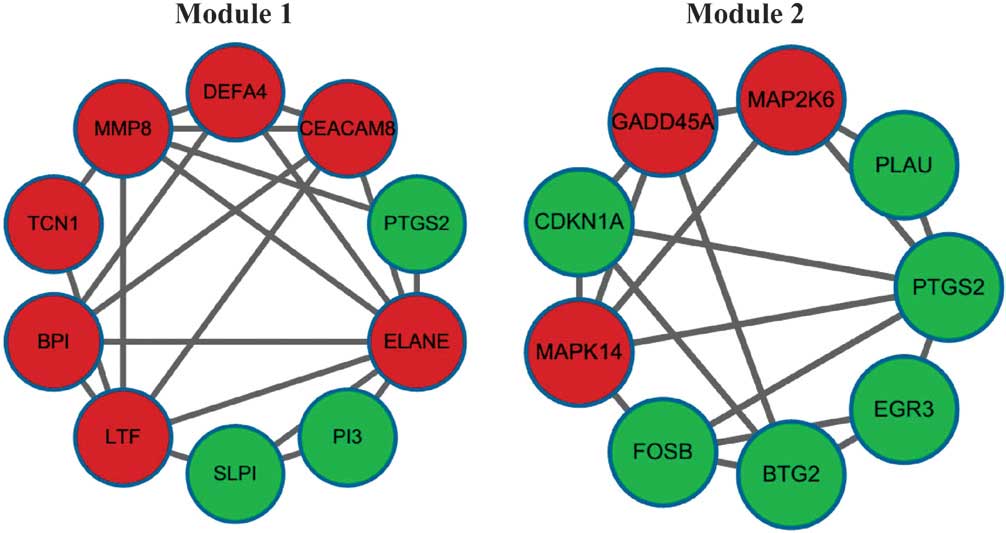
By using CFinder, three models were obtained using the default
parameter of k=3 and the top two modules are shown in
Fig. 3.
The significantly enriched GO terms and KEGG
pathways for Module 1 and Module 2 are displayed in Tables IV and V, respectively. Genes in Module 1 were
mainly classified into the GO terms of multi-organism processes,
extracellular region and serine-type endopeptidase activity, while
genes in Module 2 were mainly classified into the GO terms negative
regulation of biological processes, nucleus and protein kinase
activity. Genes in Module 2 were enriched in 12 pathways, including
the MAPK, p53 and VEGF signaling pathways, while no significant
pathway was identified for genes in Module 1 at FDR<0.05.
| Table IVGO annotation for genes in the top
two modules. |
Table IV
GO annotation for genes in the top
two modules.
| Category | GO ID | Function | Count | P-value |
|---|
| Module 1 | | | | |
| BP | GO:0050832 | Defense response to
fungus | 3 |
3.87×10−7 |
| GO:0009620 | Response to
fungus | 3 |
1.42×10−6 |
| GO:0009617 | Response to
bacterium | 5 |
1.96×10−6 |
| GO:0051704 | Multi-organism
process | 7 |
5.46×10−6 |
| GO:0051707 | Response to other
organisms | 5 |
2.53×10−5 |
| CC | GO:0005576 | Extracellular
region | 9 |
8.31×10−8 |
| GO:0031012 | Extracellular
matrix | 3 | 0.0016 |
| GO:0044421 | Extracellular
region part | 4 | 0.0031 |
| GO:0030141 | Secretory
granule | 2 | 0.0103 |
| GO:0005615 | Extracellular
space | 3 | 0.0114 |
| MF | GO:0004252 | Serine-type
endopeptidase activity | 3 |
5.05×10−5 |
| GO:0008236 | Serine-type
peptidase activity | 3 |
7.44×10−5 |
| GO:0017171 | Serine hydrolase
activity | 3 |
7.70×10−5 |
| GO:0004175 | Endopeptidase
activity | 3 | 0.0007 |
| GO:0004867 | Serine-type
endopeptidase inhibitor activity | 2 | 0.00103 |
| Module 2 | | | | |
| BP | GO:0071478 | Cellular response
to radiation | 4 |
4.48×10−7 |
| GO:2000379 | Positive regulation
of reactive oxygen species metabolic process | 3 |
9.14×10−7 |
| GO:0048519 | Negative regulation
of biological processes | 9 |
1.27×10−6 |
| GO:0009605 | Response to
external stimuli | 7 |
1.63×10−6 |
| GO:0071479 | Cellular response
to ionizing radiation | 3 |
2.02×10−6 |
| CC | GO:0005634 | Nucleus | 7 | 0.0038 |
| GO:0005654 | Nucleoplasm | 3 | 0.0219 |
| MF | GO:0004708 | Mitogen-activated
protein kinase kinase activity | 2 |
3.65×10−5 |
| GO:0004712 | Protein
serine/threonine/tyrosine kinase activity | 2 | 0.00018 |
| GO:0004672 | Protein kinase
activity | 3 | 0.0038 |
| GO:0016773 | Phosphotransferase
activity, alcohol group as acceptor | 3 | 0.0064 |
| GO:0016301 | Kinase
activity | 3 | 0.0079 |
| Table VKEGG pathway enrichment for genes in
Module 2. |
Table V
KEGG pathway enrichment for genes in
Module 2.
| KEGG ID | Function | Count | P-value |
|---|
| 4380 | Osteoclast
differentiation | 3 | 0.0005 |
| 5014 | Amyotrophic lateral
sclerosis | 2 | 0.0022 |
| 4115 | P53 signaling
pathway | 2 | 0.0035 |
| 5140 | Leishmaniasis | 2 | 0.0040 |
| 4370 | Vascular
endothelial growth factor signaling pathway | 2 | 0.0044 |
| 4010 | Mitogen-activated
protein kinase signaling pathway | 3 | 0.0044 |
| 4664 | Fc epsilon RI
signaling pathway | 2 | 0.0048 |
| 4912 |
Gonadotropin-releasing hormone signaling
pathway | 2 | 0.0077 |
| 4620 | Toll-like receptor
signaling pathway | 2 | 0.0078 |
| 4110 | Cell cycle | 2 | 0.0114 |
| 5145 | Toxoplasmosis | 2 | 0.0129 |
| 5160 | Hepatitis C | 2 | 0.0132 |
Discussion
Due to its high prevalence and mortality, there is
an urgent requirement to investigate the pathogenic mechanism of
VAP. The present study analyzed the gene expression profiles of
patients who developed VAP and those who were not affected in order
to identify possible functions and signaling pathways of DEGs
between the two groups. After the construction of a PPI network,
sub-modules in the network were mined and the functions of the top
two modules were annotated.
In the present study, a total of 69 DEGs were
identified between patients with VAP and those not affected. This
result differs from that of Swanson et al (8), who provided the gene expression
profile and identified 810 DEGs between the two groups. The
inconsistency between the present study and that by Swanson et
al (8) is likely to be
attributed to the different statistical methods employed for
detecting DEGs. Swanson et al (8) screened the DEGs using Partek software
and the analysis of variance method, which calculates a
q-value as described by Storey and Tibshirani (23), while the present study used the
Limma package in R language, which is at present most commonly
applied for identifying DEGs. To avoid false-positive results,
relatively high cutoff values of FDR<0.05 and |log (fold
change)|≥1.5 were set, which ensured high accuracy of the
results.
The upregulated genes in the VAP group identified in
the present study were mainly enriched in the GO terms of immune
system processes and immune responses, which have been verified to
have a pivotal role in the development of VAP (24). The immune system has a central role
in controlling the duration and amplitude of the inflammatory
response (25). A previous study
on sepsis revealed that the innate and adaptive immune response is
considerably different between patients with VAP and those without
this type of nosocomial infection (26). A cohort study including 90 patients
with VAP mainly caused by Gram-negative bacteria demonstrated an
association between immune system disorders and mortality, as
patients with early monocyte apoptosis of >50% were less likely
to succumb to sepsis compared with those exhibiting monocyte
apoptosis of l<50% (27). The
present study revealed that GO:0002376 (immune system processes)
and GO:0006955 (immune responses) were significantly abnormal in
patients with VAP, and the two GO categories included the genes
ELANE and LTF. ELANE is the gene encoding
neutrophil elastase, a serine protease expressed in myelomonocytic
cells and their precursors (28).
Mutations in the ELANE gene are proven to be the most common
genetic cause of congenital neutropenia, as ELANE mutations
are associated with functional deficiencies in neutrophils and
further contribute to the risk of infection (29,30).
In addition, lactoferrin, encoded by LTF, was suggested to
affect the regulation of the inflammatory response by modulating
cellular iron homeostasis (31).
Furthermore, the present study showed that ELANE and
LTF were the significant nodes in the resulting PPI network.
It is therefore inferred that ELANE and LTF may be
involved in the susceptibility of patients to VAP by affecting the
immune response.
The present study also observed that the upregulated
genes in patients with VAP were significantly enriched in the MAPK
signaling pathway, which was also the most significant pathway
enriched among genes in Module 2 mined from the PPI network. The
MAPK signaling pathway is a three-tiered cascade that includes a
MAP kinase kinase kinase (MAP3K), MAP kinase kinase (MAP2K) and
MAPK, which regulate numerous cellular functions, including
proliferation, differentiation, migration and apoptosis (32). The four major MAPKs are
extracellular regulated kinases 1 and 2 (ERK1/2), c-Jun-N-terminal
kinases, p38 and ERK5 (33).
Functional analysis performed in the present study demonstrated
that four DEGs (GADD45A, MAPK14, IL1R2 and MAP2K6)
were significantly enriched in this pathway, among which
MAPK14 is also one of the nodes with the highest connection
degree in the PPI network. MAPK14 encodes the MAPK14 protein
that is also referred to as p38α (34). p38α is the first identified member
of the p38 MAPK family and can be targeted by pyridinylimidazole
drugs that inhibit the production of pro-inflammatory cytokines
(35). Höcker et al
(36) have demonstrated that
following its activation by the GADD45B-MAP3K4 signaling complex,
MAPK14 is directed to autophagosomes, where it impairs
autophagosome-lysosome fusion and thus autophagy. It has been
indicated that activation of autophagy in macrophages mediates the
early lung inflammation during mechanical ventilation via NLRP3
inflammasome signaling (37).
These findings combined with the results of the present study lead
to the hypothesis that the p38 MAPK signaling pathway may be
involved in the development of VAP and MAPK14 may be one of
the key genes in this process.
In conclusion, the present study analyzed the gene
expression profiles between patients with VAP and those not
affected by this disease using a computational bioinformatics
approach. Functional annotation of the DEGs into GO terms and KEGG
pathways was performed, and a PPI network was constructed, followed
by module mining. Genes including ELANE, LTF and
MAPK14 may have important roles in the development of VAP
via altering the immune response and the MAPK signaling pathway.
The present study provided a novel, genetic perspective on VAP,
which may aid in the development of strategies to prevent or treat
VAP in patients in ICUs.
Acknowledgments
The present study was supported by the Nosocomial
Infection Control Research Fund of The Chinese Preventative
Association (grant no. ZHYG2014-0037) and the Jinling Hospital
Research Fund of the Jinling Affiliated Hospital of Nanjing
University Medical School (grant no. YYMS2014017).
References
|
1
|
Morehead RS and Pinto SJ:
Ventilator-associated pneumonia. Arch Intern Med. 160:1926–1936.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koenig SM and Truwit JD:
Ventilator-associated pneumonia: Diagnosis, treatment and
prevention. Clin Microbiol Rev. 19:637–657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kollef MH, Hamilton CW and Ernst FR:
Economic impact of ventilator-associated pneumonia in a large
matched cohort. Infect Control Hosp Epidemiol. 33:250–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barbier F, Andremont A, Wolff M and
Bouadma L: Hospital-acquired pneumonia and ventilator-associated
pneumonia: Recent advances in epidemiology and management. Curr
Opin Pulm Med. 19:216–228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O'Dwyer MJ, Mankan AK, Stordeur P,
O'Connell B, Duggan E, White M, Kelleher DP, McManus R and Ryan T:
The occurrence of severe sepsis and septic shock are related to
distinct patterns of cytokine gene expression. Shock. 26:544–550.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bridge DR, Novotny MJ, Moore ER and Olson
JC: Role of host cell polarity and leading edge properties in
Pseudomonas type III secretion. Microbiology. 156:356–373. 2010.
View Article : Google Scholar :
|
|
7
|
Ferrandi C, Ardissone V, Ferro P, Rückle
T, Zaratin P, Ammannati E, Hauben E, Rommel C and Cirillo R:
Phosphoinositide 3-kinase gamma inhibition plays a crucial role in
early steps of inflammation by blocking neutrophil recruitment. J
Pharmacol Exp Ther. 322:923–930. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Swanson JM, Wood GC, Xu L, Tang LE,
Meibohm B, Homayouni R, Croce MA and Fabian TC: Developing a gene
expression model for predicting ventilator-associated pneumonia in
trauma patients: A pilot study. PLoS One. 7:152012. View Article : Google Scholar
|
|
9
|
Ma J and Qin ZS: Different normalization
strategies for microarray gene expression traits affect the
heritability estimation. BMC Proc. 1(Suppl 1): S1542007. View Article : Google Scholar
|
|
10
|
Delhomme N, Padioleau I, Furlong EE and
Steinmetz LM: EasyRNASeq: A bioconductor package for processing
RNA-Seq data. Bioinformatics. 28:2532–2533. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh B, Ronghe AM, Chatterjee A, Bhat NK
and Bhat HK: MicroRNA-93 regulates NRF2 expression and is
associated with breast carcinogenesis. Carcinogenesis.
34:1165–1172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chand Y and Alam MA: Network biology
approach for identifying key regulatory genes by expression based
study of breast cancer. Bioinformation. 8:1132–1138. 2012.
View Article : Google Scholar :
|
|
13
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J R Stat Soc B (Statistical Methodology).
57:289–300. 1995.
|
|
14
|
Benjamini Y: Discovering the false
discovery rate. J R Stat Soc B (Statistical Methodology).
72:405–416. 2010. View Article : Google Scholar
|
|
15
|
Hu YH, Hu YL, Liu DW, Yu JX and Liu DM:
Screening and bioinformatics analysis of differentially expressed
genes in hyperplastic scar. Nan Fang Yi Ke Da Xue Xue Bao.
34:939–944. 2014.In Chinese. PubMed/NCBI
|
|
16
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium Nat Genet. 25:25–29. 2000.
|
|
17
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–101; discussion 101–103, 119–128, 244–252. 2002.
View Article : Google Scholar
|
|
18
|
Wingender E: The TRANSFAC project as an
example of framework technology that supports the analysis of
genomic regulation. Brief Bioinform. 9:326–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao M, Sun J and Zhao Z: TSGene: A web
resource for tumor suppressor genes. Nucleic Acids Res. 41(Database
Issue): D970–D976. 2013. View Article : Google Scholar :
|
|
20
|
Chen JS, Hung WS, Chan HH, Tsai SJ and Sun
HS: In silico identification of oncogenic potential of fyn-related
kinase in hepatocellular carcinoma. Bioinformatics. 29:420–427.
2013. View Article : Google Scholar
|
|
21
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41(Database Issue): D808–D815. 2013. View Article : Google Scholar :
|
|
22
|
Adamcsek B, Palla G, Farkas IJ, Derényi I
and Vicsek T: CFinder: Locating cliques and overlapping modules in
biological networks. Bioinformatics. 22:1021–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Storey JD and Tibshirani R: Statistical
significance for genomewide studies. Proc Natl Acad Sci USA.
100:9440–9445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gogos C, Kotsaki A, Pelekanou A,
Giannikopoulos G, Vaki I, Maravitsa P, Adamis S, Alexiou Z,
Andrianopoulos G, Antonopoulou A, et al: Early alterations of the
innate and adaptive immune statuses in sepsis according to the type
of underlying infection. Crit Care. 14:R962010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Surbatovic M, Jevdjic J, Veljovic M,
Popovic N, Djordjevic D and Radakovic S: Immune response in severe
infection: Could life-saving drugs be potentially harmful? Sci
World J. 2013:9618522013. View Article : Google Scholar
|
|
26
|
Pelekanou A, Tsangaris I, Kotsaki A,
Karagianni V, Giamarellou H, Armaganidis A and
Giamarellos-Bourboulis EJ: Decrease of CD4 -lymphocytes and
apoptosis of CD14-monocytes are characteristic alterations in
sepsis caused by ventilator-associated pneumonia: Results from an
observational study. Crit Care. 13:R1722009. View Article : Google Scholar
|
|
27
|
Giamarellos-Bourboulis EJ, Routsi C,
Plachouras D, Markaki V, Raftogiannis M, Zervakis D, Koussoulas V,
Orfanos S, Kotanidou A and Armaganidis A: Early apoptosis of blood
monocytes in the septic host: Is it a mechanism of protection in
the event of septic shock? Crit Care. 10:R762006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Germeshausen M, Deerberg S, Peter Y,
Reimer C, Kratz CP and Ballmaier M: The spectrum of ELANE mutations
and their implications in severe congenital and cyclic neutropenia.
Hum Mutat. 34:905–914. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shu Z, Li XH, Bai XM, Zhang ZY, Jiang LP,
Tang XM and Zhao XD: Clinical characteristics of severe congenital
neutropenia caused by novel ELANE gene mutations. Pediatr Infect
Dis J. 34:203–207. 2015. View Article : Google Scholar
|
|
30
|
Horwitz MS, Corey SJ, Grimes HL and
Tidwell T: ELANE mutations in cyclic and severe congenital
neutropenia: Genetics and pathophysiology. Hematol Oncol Clin North
Am. 27:19–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frioni A, Conte MP, Cutone A, Longhi C,
Musci G, di Patti MC, Natalizi T, Marazzato M, Lepanto MS, Puddu P,
et al: Lactoferrin differently modulates the inflammatory response
in epithelial models mimicking human inflammatory and infectious
diseases. Biometals. 27:843–856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang SH, Sharrocks AD and Whitmarsh AJ:
MAP kinase signalling cascades and transcriptional regulation.
Gene. 513:1–13. 2013. View Article : Google Scholar
|
|
33
|
Slattery ML, Lundgreen A and Wolff RK:
Dietary influence on MAPK-signaling pathways and risk of colon and
rectal cancer. Nutr Cancer. 65:729–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yego EC and Dillman JF III: Cytokine
regulation by MAPK activated kinase 2 in keratinocytes exposed to
sulfur mustard. Toxicol In Vitro. 27:2067–2075. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Höcker R, Walker A and Schmitz I:
Inhibition of autophagy through MAPK14-mediated phosphorylation of
ATG5. Autophagy. 9:426–428. 2013. View Article : Google Scholar :
|
|
37
|
Zhang Y, Liu G, Dull RO, Schwartz DE and
Hu G: Autophagy in pulmonary macrophages mediates lung inflammatory
injury via NLRP3 inflammasome activation during mechanical
ventilation. Am J Physiol Lung Cell Mol Physiol. 307:L173–L185.
2014. View Article : Google Scholar : PubMed/NCBI
|