Introduction
Parkinson's disease (PD) is a serious degenerative
disorder, which is the second most prevalent type of
neurodegenerative disorder in humans, particularly in older
individuals (1). A previous study
demonstrated that PD is predominantly caused by the degeneration of
dopaminergic neurons in the substantia nigra among other
localizations (2); however, the
specific cause of the cell death remains to be elucidated. Former
studies involving PD models and clinical patients have identified
two main cell death pathways, including the autophagy pathway and
the intrinsic or extrinsic apoptosis pathway (3–5).
Human postmortem studies also found that dopaminergic neuronal
death is caused by apoptosis in patients with PD (6, 7).
Fas receptor (8) and autophagic
vacuole (9) levels were observed
to be increased in patients with PD and PD experimental models
(10). Increasing evidence
supports the hypothesis that apoptosis is important in the death of
cells and degeneration of the neurons in PD.
1-Methyl-4-phenylpyridinium (MPP+) is a potential
neuronal toxin, which induces the degeneration of nigrostriatal
neurons in rodents and primates (6, 10).
Therefore, MPP+ has been used in numerous studies to
establish models of PD (6,
10, 11).
[Sar9, Met(O2)11]
termed Substance P (SP), is a widely distributed undecapeptide,
which is considered to be a neurotransmitter or neuromodulator
(12, 13) in the human central nervous system
(CNS). By interacting with the neuro-kinin-1 (NK-1) receptors, SP
is able to effectively regulate neuronal activity in a few
localizations in the brain. Thornton and Vink (14) demonstrated that SP is involved in
certain neurological diseases, such as PD and Alzheimer's disease
(AD). The electrophysiological studies of Nalivaiko et al
(15) revealed that SP could
effectively regulate nigral dopaminergic neurons (15). Strell et al (16) reported that SP could enhance
dopamine release from the striatal dopamine terminals in the brain
(16). Thus, dopamine may be
involved in maintaining the integrity of the neuronal population
(13).
In the present study, neuronal cultures were treated
with MPP+ to induce a model, and were analyzed to
evaluate the Ca2+ influx level, the caspase-3 activity,
the level of reactive oxygen species (ROS) and the mitochondrial
membrane potential (Δψm). The p38 mitogen-activated protein kinase
(MAPK) kinase and c-Jun N-terminal kinase (JNK) levels were
detected using western blotting. To investigate the protective
effects of SP, the cells were pretreated with SP prior to exposure
to MPP+. For the NK-1 receptor inhibitor (SR)
antagonism, the cells were pretreated with SR prior to treatment
with SP. For the MAPK inhibitor treatment, the cells were treated
with JNK-specifc inhibitor (SP600125) and p38-specific inhibitor
(SB203580) prior to exposure to MPP+. The aim of the
present study was to investigate whether SP could protect the
dopaminergic neurons from MPP+-triggered neurotoxicity,
and to examine the anti-apoptotic mechanism.
Materials and methods
Materials
Skimmed milk sucrose peptone was purchased from
Tocris Bioscience (Avonmouth, UK). The inhibitor, SR140333B, was
provided by Sanofi-Aventis (Chilly-Mazarin, France). Dulbecco's
modified Eagle's medium (DMEM)/F12 was purchased from Gibco Life
Technologies (Grand Island, NY, USA). The commercial phycoerythrin
(PE)-conjugated anti-caspase-3 monoclonal antibody kit was
purchased from BD Pharmingen (San Diego, CA, USA). The Hoechst
33258 staining kit was purchased from Beyotime Institute of
Biotechnology (Haimen, China). Fluo-3/AM were purchased from
Molecular Probes Life Technologies (Carlsbad, CA, USA). Rabbit
anti-phospho-P38. MAPK polyclonal antibody was purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Rabbit
anti-rat phospho-JNK polyclonal antibody and mouse anti-rat
phospho-c-Jun monoclonal antibody were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Horseradish
peroxidase (HRP)-IgG was purchased from Pierce Biotechnology, Inc.
(Rockford, IL, USA). SB20358 and SP600125 were purchased from
Molecular Probes Life Technologies. The other reagents were of the
highest grade and the other kits were purchased from local
commercial companies.
Cell culture
Dopaminergic MES23.5 cells were obtained from Dr
Wei-Dong Le (Qingdao University, Qingdao, China). MES23.5 cells
exhibit certain properties, which are similar to those of primary
neurons (17). Cells were cultured
in DM EM/F12, which was supplemented with the 5% fetal bovine
serum, 100 U/ml penicillin and 100 mg/ml streptomycin
(Sigma-Aldrich, St. Louis, MO, USA) in a humid 5% CO2
environment at 37°C. The cells were seeded at a final density of
1×105/cm2 in the plates. When the confluence
reached 70–80%, the MES23.5 cells were treated with SP (at a final
concentration of 10-7 mol/l) and SR (at a final
concentration of 10-5 mol/l) alone or in combination for
24 h. Subsequently, the cells were incubated with MPP+
(200 µmol/l) for 24 h at room temperature (18).
MTT assay
MES23.5 cells were seeded and cultured in the
96-well plates at a cell density of 2×104 cells/well.
Following cell attachment, the cells were incubated with different
doses of SP or SR for 24 h, with freshly prepared stock solutions.
Subsequently, the MPP+ (200 µmol/l) was added
into the DMEM/F12 medium and serum deprivation was conducted for
the subsequent 24 h. Subsequently, the MES23.5 cells were incubated
in MTT (5 mg/ml) for 4 h at room temperature, and cell injury was
observed using a colorimetric assay (cat. no. ab39401; Abcam,
Cambridge, UK).
Hoechst 33258 staining
The nuclear morphology of the cells was evaluated
using a previously described method (19). Briefly, the cells were fixed, then
washed twice with phosphate-buffered saline solution. Subsequently,
the cells were stained with Hoechst 33258 staining solution. The
apoptotic cells were determined by the following features: Nuclear
morphology changes, chromatin condensation and chromatin
fragmentation. Briefly, the quantity of the condensed cells was
counted manually by investigators using a fluorescence microscope
(BX51; Olympus Corporation, Tokyo, Japan) (20). The specific methods of the
experiments were performed according to those of a previous study
(19). The data are expressed as
the percentage of condensed nuclei compared with the total cell
number.
Assay of activated caspase-3
The caspase-3 activity was evaluated according to
the manufacturer's instructions (BD Pharmingen). The cells were
washed with cold PBS twice, and resuspended in Cytofix/Cytoperm™
solution (final concentration, 1×106 cells/0.5 ml; (BD
Biosciences, San Jose, CA, USA). The cells were incubated on ice
for 20 min, and were washed with Perm/wash buffer twice.
Subsequently, the cells were incubated with antibodies in the
Perm/wash buffer (1:5). Following washing with Perm/wash buffer,
the cells were resuspended with 0.5 ml Perm/wash buffer, incubated
with CD8-PE (BD Biosciences) at 37°C and analyzed using a flow
cytometric assay (FC500; Beckman Coulter, Inc., Brea, CA, USA). The
apoptosis level was evaluated by counting the caspase-3-positive
cells as a percentage of total MES23.5 cells using CellQuest
software (version 2.0; BD Biosciences).
Examination of Δψm
The Δψm of MES23.5 cells was examined by staining
with rhodamine 123 (Sigma-Aldrich), and observed using flow
cytometry (BD Biosciences). The rhodamine 123 staining and flow
cytometric observations were performed according to previously
described methods (21, 22). The cells in different groups were
incubated with 100 µmol/l ferrous iron (pH, 6.0) for 3 h at
room temperature. Subsequently, the cells were incubated with
rhodamine 123 (5 µmol/l) at 37°C for 30 min. The cells were
washed twice with HEPES-buffered saline, and the fluorescence was
observed at wavelengths of 488 nm excitation and 525 nm emission
using an inverted fluorescence microscope (IX3; Olympus
Corporation).
ROS detection
The intracellular levels of ROS were detected using
2', 7'-dichlorodihydrofuorescein 3diacetate (H2DCFDA;
Sigma-Aldrich) according to a previously described method (23). The cells were washed with PBS three
times, and were incubated with DMEM/F12 containing
H2DCFDA (10 µmol/l) for 30 min. Finally, the
fluorescence signals of the cells were observed at wavelengths of
excitation of 488 nm and emission of 525 nm.
Evaluation of intracellular
Ca2+
In the present study, intracellularfree
Ca2+, [Ca2+]i, was evaluated using
Invitrogen Fluo-3/AM (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to a previously described method (24). The cells were harvested and
centrifuged at a speed of 1,000 x g for 5 min, then treated with
Fluo-3/AM (10 µM) in serum-free DMEM for 30 min at 37°C. The
cells were analyzed and observed using the BD Biosciences FACS
Calibur flow cytometer (for Fluo-3) (25). For each experiment, 10 nM digitonin
(Sigma-Aldrich) or 2 µM ionomycin (Sigma-Aldrich) was used
to treat the cells at the end of the experiments, resulting in
maximal fluorescence of Fura-2 and Fluo-3, respectively. Finally,
the minimal fluorescence was evaluated by addition of 5 mM
EGTA.
Western blot analysis
The cells were harvested and lysed in lysis buffer
[50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 1 mM EDTA,
10 µg/ml aprotinin and 1 mM phenylmethylsulfonyl fluoride].
The nuclear and cytoplasmic proteins were extracted and isolated as
described in a previous study by Wang et al (26). The nuclear and cytoplasmic proteins
were isolated using the Nuclear and Cytoplasmic Protein Extraction
kit (Beyotime Institute of Biotechnology). The final concentration
of the proteins was examined using a Bradford assay kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The proteins were separated
with 10% SDS-PAGE (Tiangen Biotech (Beijing) Co., Ltd., Beijing,
China), and then transfer red to the polyvinylidene difluoride
membranes (Tiangen Biotech (Beijing) Co., Ltd.) for detection. The
membranes were blocked with 5% non-fat milk at room temperature
overnight, and then treated with a rabbit anti-rat phospho-P38
monoclonal antibody (1:3, 000; cat. no. S c-79 75 -R; Santa Cruz
Biotechnology, Inc.), rabbit anti-rat phospho-JNK polyclonal
antibody (1:3,000; cat. no. S c-1356 42; Santa Cruz Biotechnology,
Inc.) and rabbit antirat phospho-c-Jun polyclonal antibody
(1:2,000; cat. no. 3270; Cell Signaling Technology, Inc.),
respectively overnight at 4°C. Subsequently, the membranes were
incubated with the goat anti-rabbit polyclonal antibodies
conjugated with HRP-IgG at a dilution of 1:200 (cat. no. A27033;
Pierce Biotechnology, Inc.). The cross-reactivity of proteins was
visualized using enhanced chemiluminescence reagents (Pierce
Biotechnology, Inc.), and then analyzed via scanning densitometry
using the Tanon Image system (Tanon Science & Technology Co.,
Ltd., Shanghai, China).
Statistical analysis
All data in the present study were analyzed using
SPSS software version 19.0 (SPSS, Inc., Armonk, N Y, USA). The
results are presented as the mean ± standard error of the mean.
Each experiment was performed at least three times, and the results
were observed by at least three observers. One-way analysis of
variance followed by the Student-Newman-Keuls test was utilized to
compare the differences between the two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Immunohistochemistry
There are three subtypes of neurokinin receptors,
including NK-1, NK-2, and NK-3. The NKs have selective affinities
for SP, NKA, and NKB, respectively. It was identified that the
MES23.5 cells bear positive NK-1 expression (Fig. 1).
Cell viability of MPP+
-treated MES23.5 cells
In order to investigate whether SP exerted a
protective effect on MPP+-treated MES23.5 cells,
different doses of SP/SR alone and in combination were added to the
culture medium. When pretreated with
10−9–10−5mol/l SP for 24 h, the cell
viability was significantly increased compared with that of the
untreated cells (Table I).
However, no significant differences were identified in cell
viability among the pretreatment with SP (10−7 mol/l) +
SR (10−6 mol/l), SP (10−7 mol/l) + SR
(10−5 mol/l) and SP (10−7 mol/l) + SR
(10−4 mol/l) groups for 24 h (Table II).
 | Table IChanges in cell viability with
different doses of SP treatment. |
Table I
Changes in cell viability with
different doses of SP treatment.
| Group | MTT (% of
control) |
|---|
| Control | 100.00±3.86 |
|
MPP+ | 87.05±4.2a |
| SP
(10−9M) + MPP+ | 90.54±3.72a,b |
| SP
(10−8M + MPP+ | 91.18±3.53a,b |
| SP
(10−7M) + MPP+ | 95.03±4.73a,b |
| SP
(10−6M) + MPP+ | 93.61±5.05a,b |
| SP
(10−5M) + MPP+ | 91.96±4.02a,b |
| Table IIChanges in cell viability with
different doses of SR treatment. |
Table II
Changes in cell viability with
different doses of SR treatment.
| Group | MTT (% of
control) |
|---|
| Control | 100.00±1.33 |
|
MPP+ | 85.01±1.62a |
| SP
(10−7M) + MPP+ | 90.06±1.36a |
| SR
(10−6M) + SP (10−7M) + MPP+ | 84.94±1.74a |
| SR
(10−5M) + SP (10−7M) + MPP+ | 82.54±1.81a,b |
| SR
(10−4M) + SP (10−7M) + MPP+ | 81.49±1.64a,b |
As shown in Table I
and Table II, pretreatment with
SP (10−7 mol/l) for 24 h is able to significantly
increase the viability of the cells treated with MPP+.
Therefore, SP (10−7 mol/l) was selected to perform the
following experiments. As treatment with
10−5–10−4 mol/l SR140333B significantly
inhibited the cell viability compared with the SP
(10−7M) + MPP+ treated group, therefore,
10−5mol/l SR140333B was selected for the following
experiments.
SP inhibits caspase-3 activation in
MPP+-treated MES23.5 cells
According to a previous study, caspase-3 is a
critical protein in cell death and the apoptotic process. In the
present study, active caspase-3 was detected using the
PE-conjugated caspase-3 monoclonal antibody apoptosis kit (Fig. 2). The results indicate that the
caspase-3 activity in the MPP+ and SR + SP +
MPP+ groups was significantly h higher when compared
with the control group (Fig. 2;
P<0.05). Meanwhile, the caspase-3 activity was significantly
decreased in the SP + MPP+ group as compared with the
MPP+ group (P<0.05). Furthermore, SR treatment may
have increased the caspase-3 activity significantly, when compared
with the SP + MPP+ group alone (Fig. 2; P<0.05).
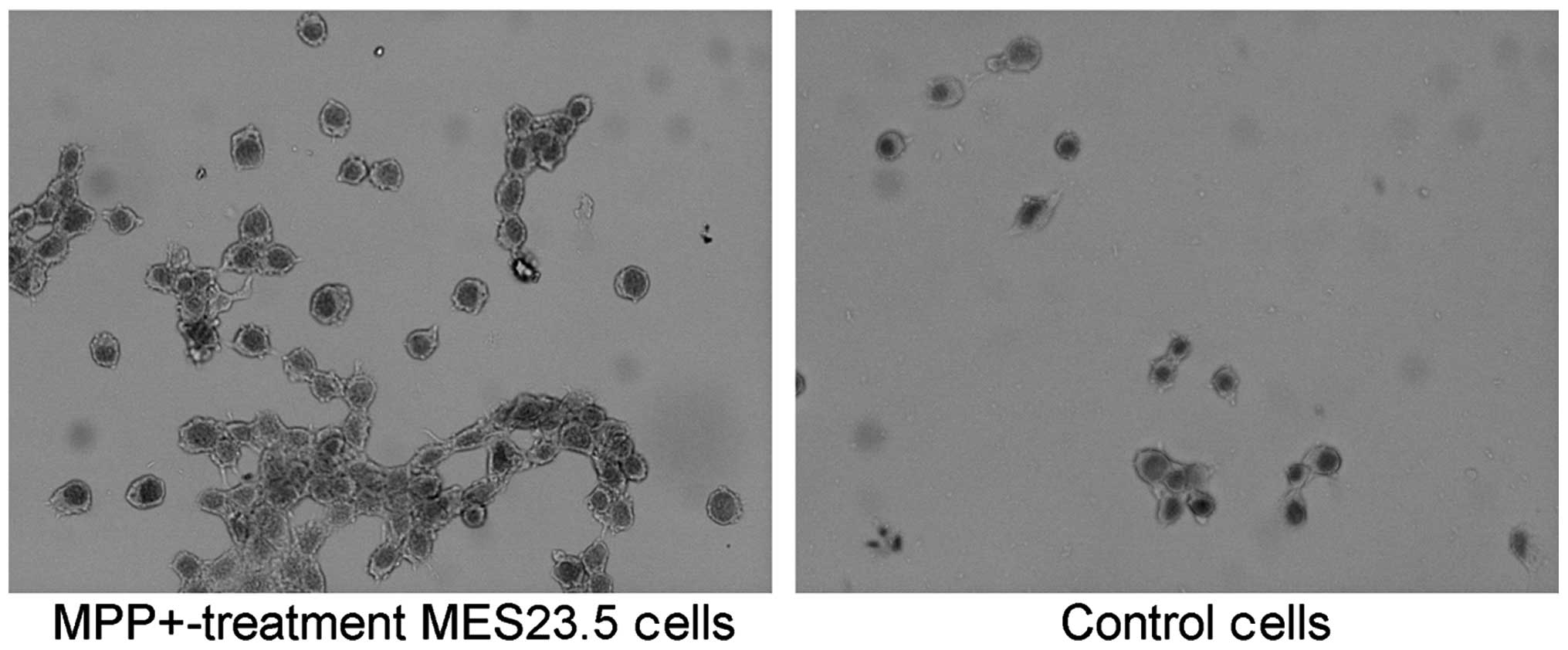
SP antagonizes DNA fragmentation in the
MPP+-treated MES23.5 cells
In order to further confirm the protective effect of
SP on MPP+-treated MES23.5 cells, the nuclear morphology
was analyzed using blue Hoechst 33258 (Fig. 3). The results demonstrate
significantly increased quantities of DNA fragmentation and
condensed nuclei in the MPP+ group when compared with
the control group (Fig. 3;
P<0.05). Treatment with SP may abrogate the increased quantities
of DNA fragmentation and condensed nuclei, as the quantity of
condensed nuclei in the SP + MPP+ group was
significantly decreased when compared with that of the
MPP+ group (P<0.05).
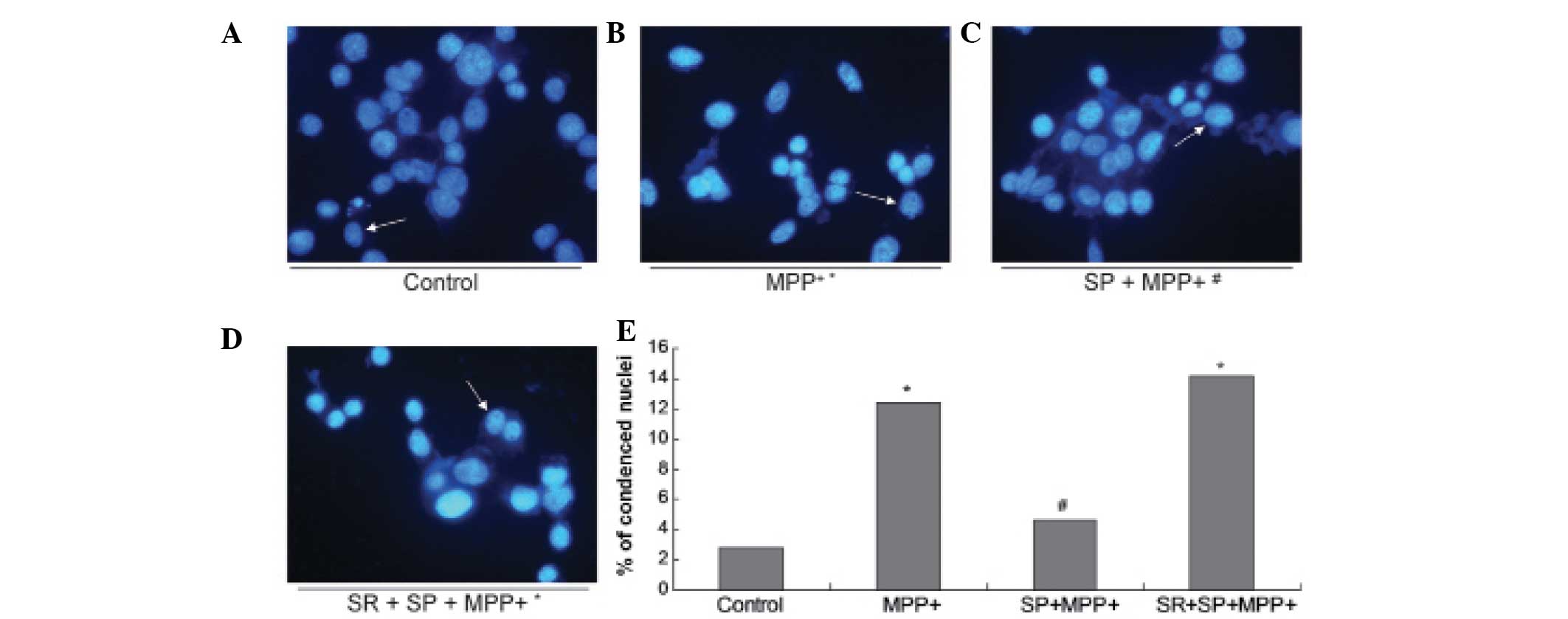 | Figure 3Morphological changes observed in the
nuclei. Images of Hoechst staining in the (A) control group, (B)
MPP+ group, (C) SP + MPP+ group and (D) SR +
SP + MPP+ group. (E) Statistical analysis of condensed
nuclei in different groups. In the control and SP groups, nuclei
appeared with regular contours and were round and large in size.
However, the nuclei of the MPP+ and SR + SP +
MPP+ groups appeared hypercondensed (brightly stained)
and exhibited fragmented chromatin. Magnification, x400. Data are
expressed as the mean ± standard error of the mean.
*P<0.05, compared with control;
#P<0.05, compared with MPP+ group.
MPP+, 1-methyl-4-phenylpyridinium; SP, substance P; SR,
NK-1 receptor inhibitor. |
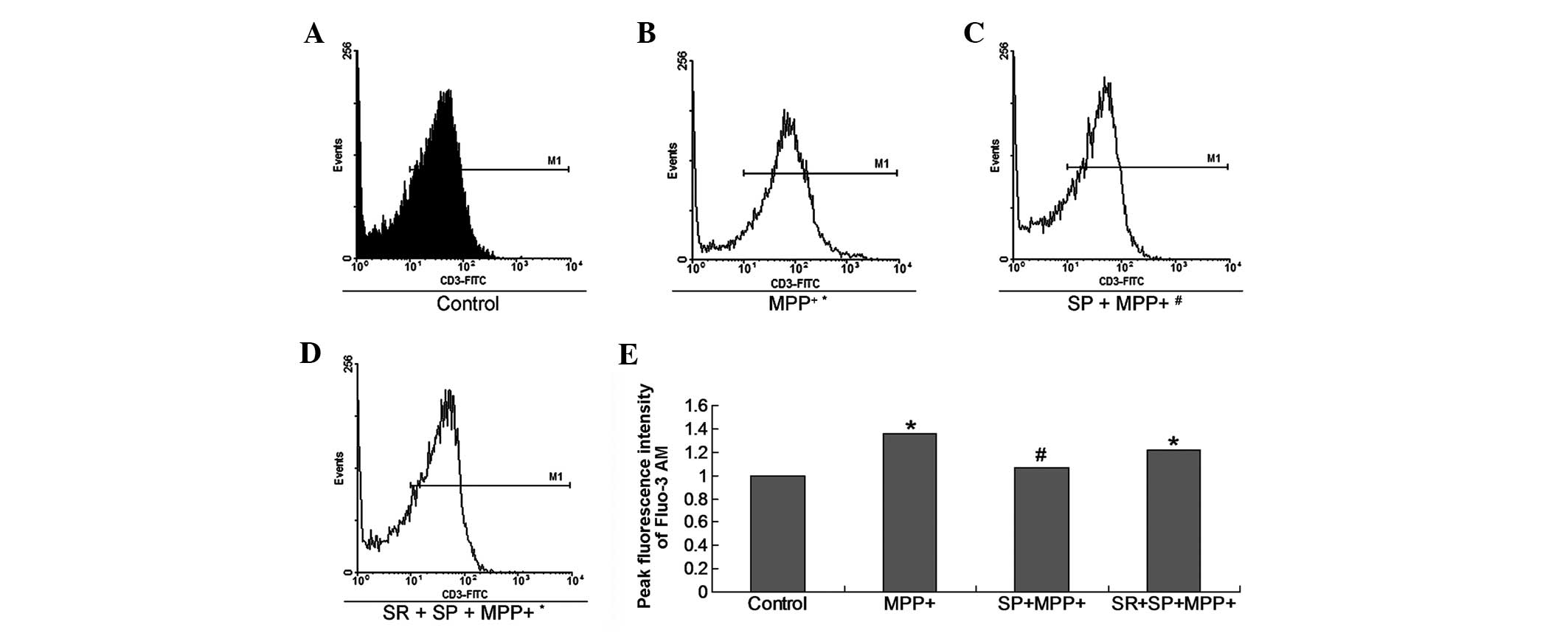
SP inhibits calcium influx in
MPP+-treated MES23.5 cells
[Ca2+]i is important in
specific cell events, such as cell apoptosis, cell death and other
processes. The Ca2+ influx measurement result indicates
that MPP+ treatment promoted Ca2+ influx in
the cultured MES23.5 cells. However, treatment with SP
significantly decreased Ca2+ influx (Fig. 4).
SP decreases ROS production in
MPP+-treated MES23.5 cells
ROS are critical in cell apoptosis, therefore,
intracellular ROS levels were detected using a fluorescence
sensitive probe, H2DCFDA, which was used to examine
numerous active oxygen species in MES23.5 cells (Fig. 5). The results indicate that the ROS
production in the MPP+ group was significantly increased
compared with the control group (Fig.
5; P<0.05). The SP treatment appeared to significantly
decrease the ROS production of the MPP+ group (Fig. 5), as the ROS production in the SP +
MPP+ group was significantly deceased when compared with
the MPP+ group (P<0.05). However, treatment with the
SR may have abrogated the effects of SP treatment (Fig. 5).
SP decreases Δψ m in
MPP+-treated MES23.5 cells
During apoptosis, the Δψm is associated with
apoptosis and represents mitochondrial function. Therefore, the Δψm
was examined in MPP+-treated MES23.5 cells (Fig. 6). The results indicate that the Δψm
in the MPP+ group was significantly increased when
compared with the control group (Fig.
6; P<0.05). The SP treatment may also significantly decrease
the Δψm of the MPP+ group (Fig. 6). Furthermore, the Δψm in the SP +
MPP+ group was significantly deceased when compared with the
MPP+ group (P<0.05). However, treatment with the SR
may have abrogated the effects of SP treatment (Fig. 6).
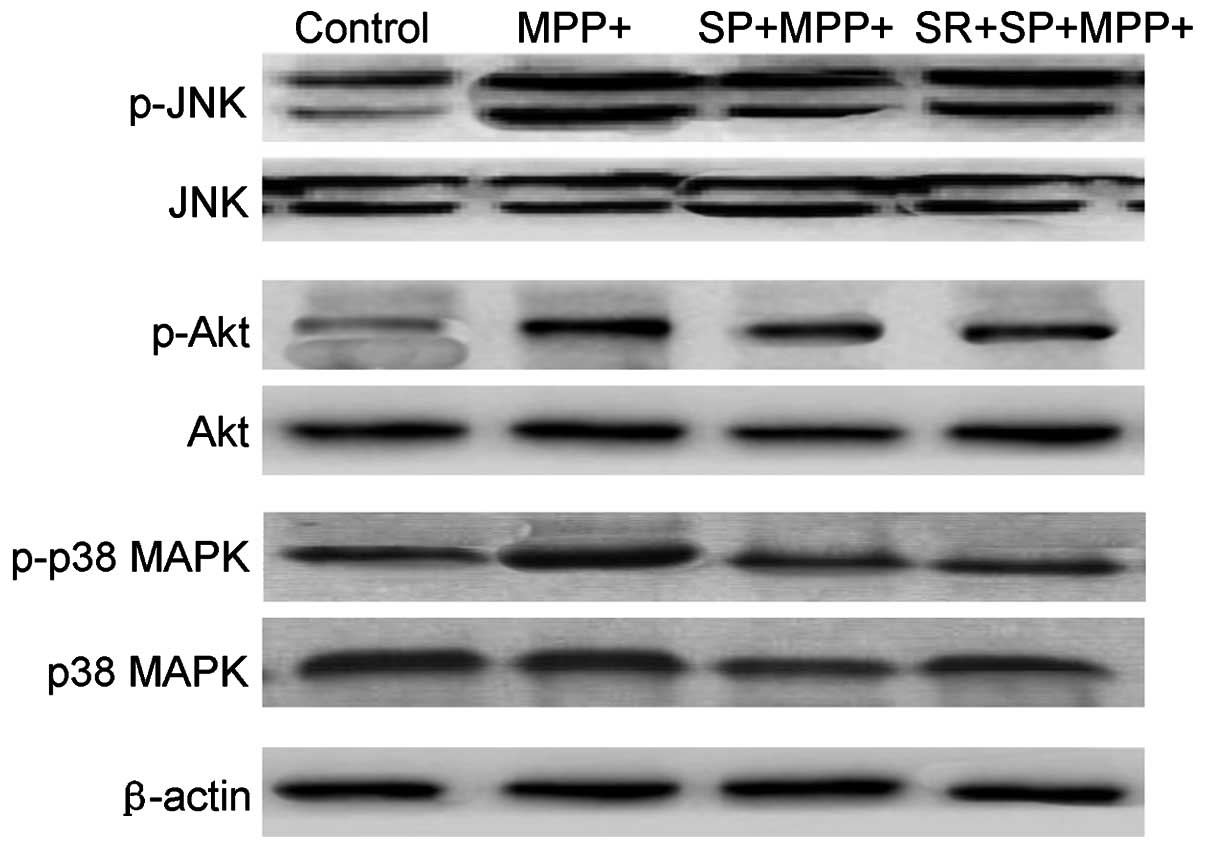
SP inhibits the MMP+ -induced
phosphorylation of JNK, p38 MAPK and Akt
Fig. 7 demonstrates
that MMP+ treatment may lead to a marginal increase in
the phosphorylation of JNK, p38 MAPK and Akt in MES23.5 cells
without altering the total level of the above three proteins.
However, treatment with SP significantly inhibited the activation,
also termed phosphorylation, of p38 MAPK, JNK and Akt protein
(Fig. 7). The present results also
revealed that SP treatment could regulate the activation of JNK,
p38 and Akt protein in MMP+-injured MES23.5 cells.
Discussion
Previous studies have reported that SP is critical
in PD pathophysiology (12,
15, 16). However, the mechanism underlying
the function of SP-induced neuroprotection in PD remains elusive. A
previous study only demonstrated the neural growth function of SP
(27). The present study indicated
the neuroprotective function of SP in the MPP+-treated
MES23.5 cells, and further examined the underlying mechanism of
this effect. The present study demonstrated that SP may inhibit the
MPP+-triggered neurotoxicity through inhibiting cell
apoptosis via the NK-1 receptor in MES23.5 cells. Furthermore, the
neuroprotective effects of SP were achieved by regulating the Δψm,
changing the calcium influx, and modulating the ROS production and
caspase-3 activation.
In the present study, the MES23.5 cell line was
selected mainly due to evidence suggesting that it contains certain
neuronal characteristics, including a dopamine synthesis system,
tyrosine hydroxylase and expression of the ω-conotoxin receptor
(28). The above characteristics
are similar to the features of primary neurons. Therefore, the
results obtained from this cell line may be used to represent the
degenerated neurons in PD.
MPP+ is a common type of neurotoxic agent
used to establish models of PD. MPP+ may be transduced
into the cell via the cells dopamine re-uptake system.
Subsequently, MPP+ is able to inhibit the formation of
complex I in the mitochondrial respiratory chain, which may trigger
dopaminergic neurode-generation (29).
The etiology of PD remains to be fully determined.
Apoptosis-associated neuronal death has only been described in a
few patients with PD (30).
Consistent with the previous study (30), the morphological data obtained in
the present study demonstrated that dopaminergic cell death indeed
occurs by triggering apoptosis in a model of PD.
Increasing numbers of studies have clearly
demonstrated the function of apoptosis in neuronal death and
neurodegeneration of dopaminergic neurons in patients with PD
(31, 32). These studies have also indicated
the critical role of the caspase family of cysteine proteases in
apoptosis. These enzymes are involved in the cascade, which is
triggered in response to pro-apoptotic signals and culminates in
the cleavage of a set of proteins (33). In the present study, the occurrence
of apoptosis was evaluated by examining caspase-3 activation
(cleaved caspase-3 protein).
There are multiple studies demonstrating that ROS
are involved in neurodegenerative diseases, such as AD and PD
(23, 26, 30,
31). Certain studies have also
illustrated that the ROS could lead to cell death via the
activation of the cell apoptosis signaling pathway (26, 30). In the present study, the results
indicated that MPP+ treatment could induce an increased
level of ROS in MES23.5 cells. However, this increase in ROS could
be blocked by pretreatment with SP. These results suggest that the
anti-apoptotic effects of SP on MPP+-treated MES23.5
cells were achieved by triggering the mitochondrial pathway.
In addition, CNS tissues have been analyzed (which
were derived from patients with PD, Huntington's disease and
amyotrophic lateral sclerosis) in order to examine the role of
cellular Ca2+ in the neurons. The results indicated that
the cellular Ca2+ overload is the direct cause for the
death of vulnerable neurons in the aforementioned diseases
(24, 30). Beal (34) demonstrated that excessive
Ca2+-mediated nitric oxide production contributes to the
death of dopaminergic neurons in PD (34). Furthermore, Ca2+ is also
one of the most critical signaling molecules in mammalian cells.
Ca2+ may also regulate the diverse cellular functions of
cells (35). Therefore, the rise
of intracellular Ca2+ may trigger neuronal cell death or
apoptosis in PD (36). The
association between [Ca2+]er and cell death
is rather complex. There is a substantial body of data
demonstrating that reducing [Ca2+]er has a
protective effect against apoptosis, dependent on stimuli releasing
Ca2+ from intracellular stores (37).
The JNK signaling pathway is a major transduction
pathway, which may mediate apoptotic cell death, or even neuronal
degeneration in response to numerous cellular stimuli (38). JNK is a member of a subfamily of
the MAPK superfamily. JNK acts as a pro-apoptotic kinase in the
apoptotic processes, including the extrinsic apoptotic pathway and
the intrinsic apoptotic pathways. Therefore, JNK activates the
apoptotic signaling pathway by upregulating the pro-apoptotic
proteins. The regulation was completed by phosphorylating specific
transcription factors, such as ATF2, c-Jun, p53, Elk-1 and c-Myc.
In addition, JNK could also modulate the activities of anti- and
pro-apoptotic proteins directly, such as B-cell lymphoma 2 and
Bcl-2-associated X protein (39).
SP600125 is a common JNK inhibitor, which could inhibit the
phosphorylation of c-Jun and protect against transient
ischemia/reperfusion-triggered neuronal death or apoptosis in the
hippocampal CA1 region of the rat. SP600125, at a concentration of
2–10 µm, may markedly protected the
cortical neurons from glutamate toxicity (40). In the present study, the results
indicated that SP inhibited the activation of JNK in
MPP+-injured MES23.5 cells. However, SP also fails to
rescue the glutamate-injured hippocampal neurons from cell
apoptosis in the presence of SP600125. A previous study revealed
that SP600125 is able to regulate Akt phosphorylation when serum
and potassium were withdrawn in the cerebellar granule cells
(41). However, in the present
MPP+ injury model of PD, the results indicated that the
SP600125-induced JNK inhibition could not activate the Akt protein
(data not shown). Therefore, this phenomenon also requires further
clarification.
P38 is another important member of the MAPK protein
family, which may be activated in response to glutamate stimulation
of the N-methyl-D-aspartate receptors (42). SB203580 is a type of
p38MAPK-specifc inhibitor, which may prevent glutamate-induced
neuronal death and apoptosis in cultured cortical neurons (43). In the present study, the results
revealed that MPP+ could detectably stimulate the
phosphorylation of p38 in MES23.5 cells. In addition, SP may affect
p38 activation, which indicates that p38 is involved in the
neuroprotective function of SP in the MPP+-injured
MES23.5 cells.
In conclusion, the present study demonstrated the
antioxidant effects of SP against MPP+-induced cell
death and apoptosis, by decreasing calcium influx, increasing the
Δψm, reducing the ROS production and inhibiting caspase-3
activation. The phosphorylation of the p38 protein and JNK protein
signaling pathways were regulated by treatment with SP The
SP-activated p38 and JNK signaling pathway promoted the cell
viability and survival of cells. For clinical therapy, SP may be
developed as a potential drug for treating neuronal cell damage and
neurodegenerative disorders, based on its neuroprotective
function.
Acknowledgments
The authors would like to thank Dr Wei-Dong Le
(Qingdao University, Qingdao, China) for providing the MES23.5 cell
line and thank Sanofi-Aventis (Chilly-Mazarin, France) for
providing the SR140333B. This study was supported by a grant from
the National Natural Science Foundation of China (grant nos.
31070942 and 81200872) and the Doctoral Fund of Ministry of
Education of China (grant no. 20123706110001).
References
|
1
|
Li DW, Liu ZQ, Chen W, Yao M and Li GR:
Association of glycogen synthase kinase-3β with Parkinson's disease
(review). Mol Med Rep. 9:2043–2050. 2014.PubMed/NCBI
|
|
2
|
Obeso JA, Rodríguez-Oroz MC, Rodríguez M,
Lanciego JL, Artieda J, Gonzalo N and Olanow CW: Pathophysiology of
the basal ganglia in Parkinson's disease. Trends Neurosci.
23(Suppl): S8–S19. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oertel WH and Ellgring H: Parkinson's
disease - medical education and psychosocial aspects. Patient Educ
Couns. 26:71–79. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levy OA, Malagelada C and Greene LA: Cell
death pathways in Parkinson's disease: Proximal triggers, distal
effectors, and final steps. Apoptosis. 14:478–500. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Irrcher I and Park DS: Parkinson's
disease: To live or die by autophagy. Sci Signal. 2:pe212009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hartmann A and Hirsch EC: Parkinson's
disease. The apoptosis hypothesis revisited. Adv Neurol.
86:143–153. 2001.PubMed/NCBI
|
|
7
|
Tatton WG, Chalmers-Redman R, Brown D and
Tatton N: Apoptosis in Parkinson's disease: Signals for neuronal
degradation. Ann Neurol. 53(Suppl 3): S61–S72. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chang WA, Lin ES, Tsai MJ, Huang MS and
Kuo PL: Isolinderalactone inhibits proliferation of A549 human non
small cell lung cancer cells by arresting the cell cycle at the
G0/G1 phase and inducing a Fas receptor and soluble Fas
ligand-mediated apoptotic pathway. Mol Med Rep. 9:1653–1659.
2014.PubMed/NCBI
|
|
9
|
Anglade P, Vyas S, Javoy-Agid F, Herrero
MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC and
Agid Y: Apoptosis and autophagy in nigral neurons of patients with
Parkinson's disease. Histol Histopathol. 12:25–31. 1997.PubMed/NCBI
|
|
10
|
Fornai F, Lenzi P, Gesi M, Soldani P,
Ferrucci M, Lazzeri G, Capobianco L, Battaglia G, De Blasi A,
Nicoletti F, et al: Methamphetamine produces neuronal inclusions in
the nigrostriatal system and in PC12 cells. J Neurochem.
88:114–123. 2004. View Article : Google Scholar
|
|
11
|
Yakhine-Diop SM, Bravo-San Pedro JM,
Gómez-Sánchez R, Pizarro-Estrella E, Rodríguez-Arribas M, Climent
V, Aiastui A, López de Munain A, Fuentes JM and González-Polo RA:
G2019S LRRK2 mutant fibroblasts from Parkinson's disease patients
show increased sensitivity to neurotoxin
1-methyl-4-phenylpyridinium dependent of autophagy. Toxicology.
324:1–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robinson P, Garza A, Moore J, Eckols TK,
Parti S, Balaji V, Vallejo J and Tweardy DJ: Substance P is
required for the pathogenesis of EMCV infection in mice. Int J Clin
Exp Med. 2:76–86. 2009.PubMed/NCBI
|
|
13
|
Barker R: Tachykinins, neurotrophism and
neurodegenerative diseases: A critical review on the possible role
of tachykinins in the aetiology of CNS diseases. Rev Neurosci.
7:187–214. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thornton E and Vink R: Treatment with a
substance P receptor antagonist is neuroprotective in the
intrastriatal 6-hydroxydopamine model of early Parkinson's disease.
PLoS One. 7:e341382012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nalivaiko E, Michaud JC, Soubrié P, Le Fur
G and Feltz P: Tachykinin neurokinin-1 and neurokinin-3
receptor-mediated responses in guinea-pig substantia nigra: An in
vitro electrophysiological study. Neuroscience. 78:745–757. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strell C, Sievers A, Bastian P, Lang K,
Niggemann B, Zänker KS and Entschladen F: Divergent effects of
norepinephrine, dopamine and substance P on the activation,
differentiation and effector functions of human cytotoxic T
lymphocytes. BMC Immunol. 10:622009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crawford GD Jr, Le WD, Smith RG, Xie WJ,
Stefani E and Appel SH: A novel N18TG2 x mesencephalon cell hybrid
expresses properties that suggest a dopaminergic cell line of
substantia nigra origin. J Neurosci. 12:3392–3398. 1992.PubMed/NCBI
|
|
18
|
Liu L, Xu H, Jiang H, Wang J, Song N and
Xie J: Ghrelin prevents 1-methyl-4-phenylpyridinium ion-induced
cytotoxicity through antioxidation and NF-kappaB modulation in
MES23.5 cells. Exp Neurol. 222:25–29. 2010. View Article : Google Scholar
|
|
19
|
Yao G, Yang L, Hu Y, Liang J, Liang J and
Hou Y: Nonylphenol-induced thymocyte apoptosis involved caspase-3
activation and mitochondrial depolarization. Mol Immunol.
43:915–926. 2006. View Article : Google Scholar
|
|
20
|
Feng Z and Zhang JT: Protective effect of
melatonin on b-amyloid induced apoptosis in rat astroglioma c6
cells and its mechanism. Free Radic Biol Med. 2004:1790–1801.
2014.
|
|
21
|
Wei CD, Li Y, Zheng HY, Tong YQ and Dai W:
Palmitate induces H9c2 cell apoptosis by increasing reactive oxygen
species generation and activation of the ERK1/2 signaling pathway.
Mol Med Rep. 7:855–861. 2013.PubMed/NCBI
|
|
22
|
Zhao L, Teng B, Wen L, Feng Q, Wang H, Li
N, Wang Y and Liang Z: mTOR inhibitor AZD8055 inhibits
proliferation and induces apoptosis in laryngeal carcinoma. Int J
Clin Exp Med. 7:337–347. 2014.PubMed/NCBI
|
|
23
|
Egnatchik RA, Leamy AK, Jacobson DA,
Shiota M and Young JD: ER calcium release promotes mitochondrial
dysfunction and hepatic cell lipotoxicity in response to palmitate
overload. Mol Metab. 3:544–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang XJ and Xu JX: Possible involvement of
Ca2+ signaling in rotenone-induced apoptosis in human
neuroblastoma SH-SY5Y cells. Neurosci Lett. 376:127–132. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bhattacharyya S, Ghosh S, Shant J, Ganguly
NK and Majumdar S: Role of the W07-toxin on Vibrio cholerae-induced
diarrhoea. Biochim Biophys Acta. 1670:69–80. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Du XX, Jiang H and Xie JX:
Curcumin attenuates 6-hydroxydopamine-induced cytotoxicity by
anti-oxidation and nuclear factor-kappa B modulation in MES23.5
cells. Biochem Pharmacol. 78:178–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iwasaki Y, Kinoshita M, Ikeda K, Takamiya
K and Shiojima T: Trophic effect of various neuropeptides on the
cultured ventral spinal cord of rat embryo. Neurosci Lett.
101:316–320. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bleiblo F, Michael P, Brabant D, Ramana
CV, Tai T, Saleh M, Parrillo JE and Kumar A and Kumar A: JAK
kinases are required for the bacterial RNA and poly I:C induced
tyrosine phosphorylation of PKR. Int J Clin Exp Med. 6:16–25.
2013.
|
|
29
|
Kitamura Y, Shimohama S, Akaike A and
Taniguchi T: The parkinsonian models: Invertebrates to mammals. Jpn
J Pharmacol. 84:237–243. 2000. View Article : Google Scholar
|
|
30
|
Siddique YH, Naz F and Jyoti S: Effect of
curcumin on lifespan, activity pattern, oxidative stress, and
apoptosis in the brains of transgenic Drosophila model of
Parkinson's disease. BioMed Res Int. 606928:20142014.
|
|
31
|
Zhang Z, Zhang K, Du X and Li Y:
Neuroprotection of desferrioxamine in lipopolysaccharide-induced
nigrostriatal dopamine neuron degeneration. Mol Med Rep. 5:562–566.
2012.
|
|
32
|
Ekshyyan O and Aw TY: Apoptosis: A key in
neurodegenerative disorders. Curr Neurovasc Res. 1:355–371. 2004.
View Article : Google Scholar
|
|
33
|
Enari M, Sakahira H, Yokoyama H, Okawa K,
Iwamatsu A and Nagata S: A caspase-activated DNase that degrades
DNA during apoptosis, and its inhibitor ICAD. Nature. 391:43–50.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Beal MF: Excitotoxicity and nitric oxide
in Parkinson's disease pathogenesis. Ann Neurol. 44(Suppl 1):
S110–S114. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ma S, Cai C, Ma Y, Bai Z, Meng X, Yang X,
Zou F and Ge R: Store-operated Ca2+ entry
mediated regulation of polarization in differentiated human
neutrophil-like HL-60 cells under hypoxia. Mol Med Rep. 9:819–824.
2014.PubMed/NCBI
|
|
36
|
Kim HY, LaVaute T, Iwai K, Klausner RD and
Rouault TA: Identification of a conserved and functional
iron-responsive element in the 5'-untranslated region of mammalian
mitochondrial aconitase. J Biol Chem. 271:24226–24230. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scorrano L, Oakes SA, Opferman JT, Cheng
EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ: BAX and BAK
regulation of endoplasmic reticulum Ca2+: A control
point for apoptosis. Science. 300:135–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brecht S, Kirchhof R, Chromik A, Willesen
M, Nicolaus T, Raivich G, Wessig J, Waetzig V, Goetz M, Claussen M,
et al: Specific pathophysiological functions of JNK isoforms in the
brain. Eur J Neurosci. 21:363–377. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Eminel S, Roemer L, Waetzig V and Herdegen
T: c-Jun N-terminal kinases trigger both degeneration and neurite
outgrowth in primary hippocampal and cortical neurons. J Neurochem.
104:957–969. 2008. View Article : Google Scholar
|
|
41
|
Yeste-Velasco M, Folch J, Casadesús G,
Smith MA, Pallàs M and Camins A: Neuroprotection by c-Jun
NH2-terminal kinase inhibitor SP600125 against potassium
deprivation-induced apoptosis involves the Akt pathway and
inhibition of cell cycle reentry. Neuroscience. 159:1135–1147.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsu MJ, Hsu CY, Chen BC, Chen MC, Ou G and
Lin CH: Apoptosis signal-regulating kinase 1 in amyloid beta
peptide-induced cerebral endothelial cell apoptosis. J Neurosci.
27:5719–5729. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu XW, Ji EF, He P, Xing RX, Tian BX and
Li XD: Protective effects of the p38 MAPK inhibitor SB203580 on
NMDA induced injury in primary cerebral cortical neurons. Mol Med
Rep. 10:1942–1948. 2014.PubMed/NCBI
|