Introduction
Chronic myelogenous leukemia (CML) is the most
common type of malignant cancer of the myeloproliferative
neoplasms. CML accounts for ~20% of all cases of leukemia in the
global population (1). Numerous
previous studies (2–4) demonstrated that genes and microRNAs
(miRs) have various roles in CML. Dysregulated genes and miRs are
important in the pathogenesis of CML, for example, the tumor
protein 63 (TP63) mutation may function as a gene alteration
and, thus, be responsible for the development of CML (5). Furthermore, downregulation of miR-10a
may increase upstream stimulatory factor 2 expression levels and
contribute to the increase in cell proliferation in CML (6). Genes and miRs that are associated
with CML, but are non-dysregulated also have a role in CML, for
example, runt-related transcription factor 3 (RUNX3) is
involved in CML persistence despite imatinib treatment (7) and miR-30a may inhibit the
effectiveness of imatinib-mediated apoptosis in CML (8).
Gene regulatory factors are predominantly
transcription factors (TFs) and microRNAs (miRs), which control the
expression of genomic information in multicellular genomes
(9). TFs are proteins that bind to
specific DNA sequences, controlling the encoding of genetic
information from DNA to messenger RNA (10). TFs, alone or with other proteins,
regulate gene expression by activation or suppression. miR is a
small non-coding RNA molecule (~22 nt in length), which functions
in post-transcriptional regulation of gene expression and is also
involved in transcriptional regulation (11). miRs regulate gene expression via
silencing or target degradation and affect various cancer
processes, including proliferation, differentiation and
apoptosis.
miRs target thousands of genes, called target genes,
and these are important for investigating the biological function
of miRs. There are many methods to evaluate the regulatory
associations between miRs and their target genes, such as
microRNA.org (12),
TargetScan (13) and PicTar
(14). There are also
experimentally validated databases, such as TarBase (15) miRTarBase (16) and miRecords (17), which provide abundant data to
evaluate the associations between miRs and genes.
miRs may be encoded in the DNA sequence of a gene,
known as a host gene. During the transcription process, miRs and
their host gene are transcribed simultaneously (18). The host gene and its intronic miR
are often involved in the same biological processes (19). The two perform certain functions
and are involved in signaling pathways (20).
The onset of CML may result from numerous
dysregulated genes and miRs. Many genes (21) and miRs (22) associated with CML have been
identified; however, the underlying mechanisms in CML remain
unclear. The present study focused on the network of TFs, miRs, and
their target and host genes, to establish the key pathways of CML
and partly indicate their control mechanisms in CML. Experimentally
validated regulatory associations (TFs→miRs, miRs→target genes, and
host genes→miRs) were collected from known databases (15–17,23).
Dysregulated genes and miRs, as well as CML-associated genes and
miRs were collected from known databases and the PubMed database
(http://www.ncbi.nlm.nih.gov/pubmed/).
To further investigate the transcription process of CML, TFs were
selected by the P-Match method (24) and considered as CML-associated
genes. Three networks were constructed to demonstrate the
regulatory mechanism of CML. The first network is the network of
miRs and genes from experimentally validated data. The second
network is the dysregulatory network, which consists of
dysregulatory data (genes and miRs) and their regulatory
associations from the first network. The third is the
CML-associated network, which consists of CML-associated data
(genes and miRs) with their regulatory associations from the first
network. In the current study, regulatory associations are referred
to as pathways. To allow comparison of the similarities and
differences distinguishing the key pathways in CML, the
dysregulated gene and miR pathways, and predicted TFs were
extracted from the three networks separately. These pathways of
dysregulated genes and miRs may be particularly important in the
development of CML (25), as the
dysregulatory network contributes to the pathogenesis of CML (with
regard to genes and miRs) while the associated network describes
the regulatory mechanisms of CML.
Materials and methods
Material collection and data
processing
An experimentally validated dataset of miRs and
their target genes were collected from Tarbase version 5.0
(15), miRTarBase version 3.5
(16) and miRecords version 4
(17). The National Center for
Biotechnology Information (NCBI) gene database (http://www.ncbi.nlm.nih.gov/gene/) was used to
unify official symbols of miRs and genes. This dataset was
designated as set U1. An experimentally validated
dataset between TFs and miRs was collected from TransmiR version
1.2 (23). This dataset was
designated as set U2. A dataset of host genes and their
miRs was collected from the NCBI gene database, and this dataset
was designated as set U3.
In the present study, the dysregulated genes include
genes with mutations, abnormal expression and single nucleotide
polymorphisms, and inactivated, overexpressed, underexpressed,
downregulated, upregulated and differentially expressed genes. The
dataset of dysregulated genes was collected from Cancer Genetics
Web (http://www.cancerindex.org/geneweb/index.html), the
KEGG pathway database (26) and
the PubMed database. CML-associated genes include dysregulated
genes, and genes associated with prevention and radial therapy
(27,28). A dataset of CML-associated genes
was collected from the GeneCards database (29), the PubMed database and included
dysregulated genes. To improve understanding of the transcriptional
network involving TFs, miRs and targets genes, predicted TFs were
extracted using the P-Match method (24). These TFs are suggested as
CML-associated genes. In the present study, TFs that appear in
TransmiR were focused on. Promoter region sequences (1,000 nt) of
target genes that are targeted by dysregulated miRs were downloaded
from University of California, Santa Cruz, Genome Browser (30). The P-Match method was used to
identify TF binding sites (TFBSs) in the 1,000-nt promoter region
sequences and the TFBSs were mapped onto the 1,000-nt promoter
region of target genes; the corresponding TFs were obtained using
these TFBSs. Matrix libraries of P-Match data, in addition to sets
of known TF-binding sites collected in TRANSFAC®
(31) enabled the identification
of a large variety of TF binding sites. The vertebrate matrix was
used with restricted high quality criterion for the matrix. The
dataset of dysregulated genes was designated as set U4
and the associated genes were designated as set U5.
Dysregulated miRs include deletions, mutations, and
differential, overexpressed, low expression, and down- and
upregulated miRs. The dataset of dysregulated miRs was collected
from the PubMed database. This dataset was designated as set
U6. The associated miRs are involved in various CML
processes and include dysregulated and non-dysregulated miRs. The
dataset of associated miRs were collected from the PubMed database
and the HMDD database 2.0 (32).
This dataset was designated as set U7.
Construction of the three networks
Experimentally validated, dysregulatory and
associated networks were constructed. Regulatory associations of
TFs, miRs, target and host genes were extracted from sets
U1, U2 and U3, these nodes were
combined and their associations used to construct the
experimentally validated network. Dysregulated genes and miRs were
extracted from sets U4 and U6, and mapped
onto the experimentally validated network. These genes, miRs and
host genes, as well as their associations, were constructed into
the dysregulatory network using the above data and other
dysregulated data (genes and miRs), which do not have regulatory
associations with other genes and miRs.
A similar method was used to construct the
associated network. Associated genes and miRs were extracted from
sets U5 and U6, and were mapped onto an
experimentally validated network. These genes, miRs, host genes and
their associations were extracted, and the associated network was
constructed using the above data and other associated data (genes
and miRs), which do not have regulatory associations with other
genes and miRs.
Results
Dysregulatory network of CML
Dysregulated miRs and genes may result in the onset
of CML. Fig. 1 presents the
dysregulatory network of CML. This network includes 31 genes, 20
miRs, 50 of their regulatory associations, 89 single nodes (genes)
and four single nodes (miRs). Fig.
1 presents various types of regulatory associations between
miRs and genes: One miR may target one or numerous genes, one TF
may regulate one or numerous miRs, several TFs may regulate one or
numerous miRs and numerous miRs may target one or numerous genes.
This dysregulatory network partly demonstrates the regulatory
mechanism of CML.
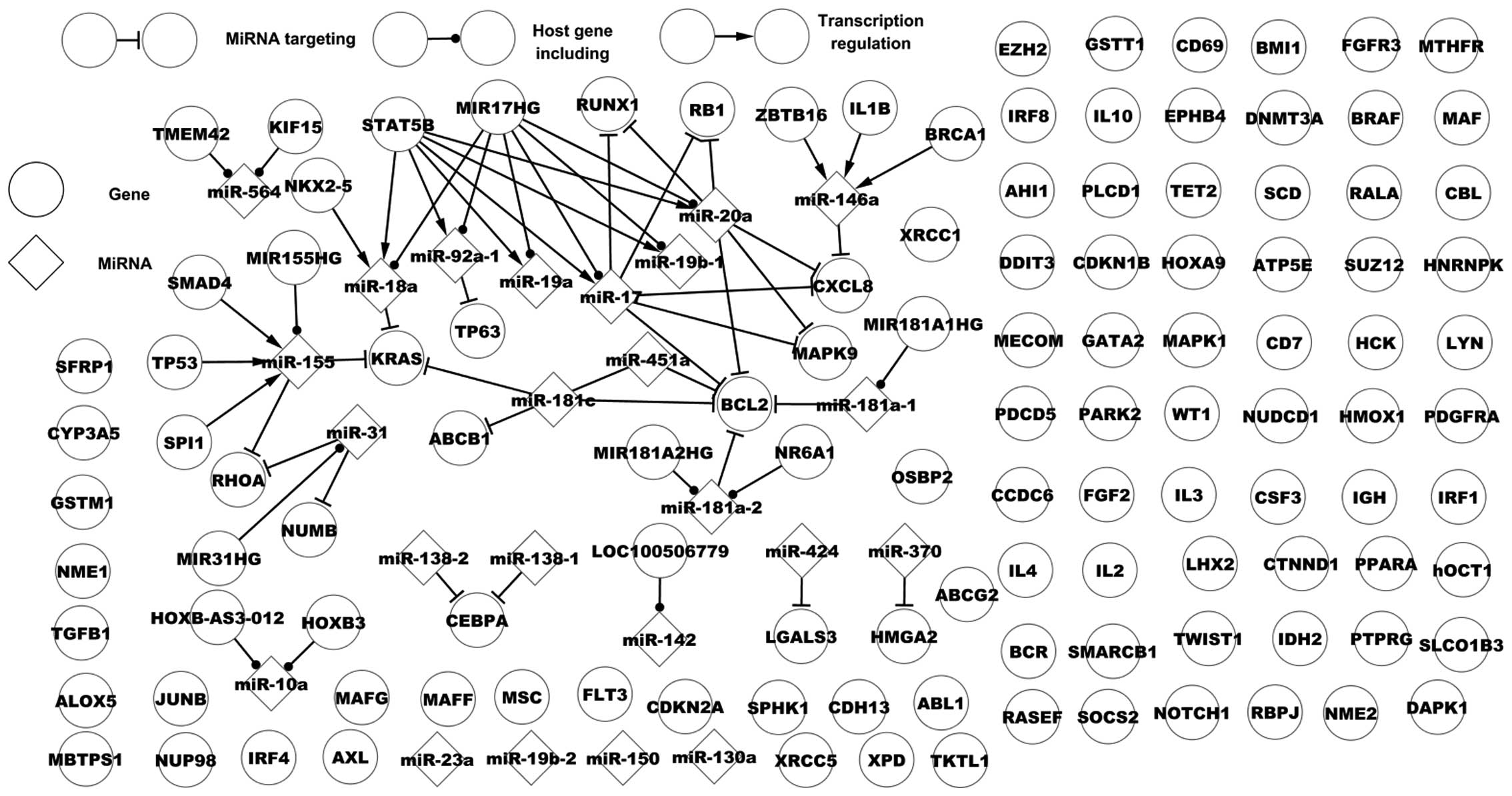 | Figure 1Dysregulatory network of chronic
myelogenous leukemia. TMEM42, transmembrane protein 42;
KIF15, kinesin family member 15; STAT5B, signal
transducer and activator of transcription 5B; MIR17HG,
miR-17–92 cluster host gene; RUNX1, runt-related
transcription factor 1; RB1, retinoblastoma 1;
NKX2-5, NK2 homeobox 5; IL8, interleukin-8;
TP63, tumor protein 63; MIR181A1HG, MIR181A1 host
gene; MAPK9, mitogen-activated protein kinase 9;
TP53, tumor protein 53; KRAS, Kirsten rat sarcoma
viral oncogene homolog; BCL2, B-cell CLL/lymphoma 2;
SPI1, Spi-1 proto-oncogene. |
Certain features of host genes and their miRs are
presented in Fig. 1. A host gene
includes one miR, which targets a number of genes; for example,
MIR181A1 host gene (MIR181A1HG) includes miR-181a-1 that
targets B-cell CLL/lymphoma 2 (BCL2). One host gene may
include many miRs with targets. One miR may be located in various
genes, for example, transmembrane protein 42 and kinesin family
member 15 include miR-564. Fig. 1
presents a host gene that includes multiple miRs that alone or
together target a number of genes. A notable host gene, MIR17HG,
contains six miRs, miR-20a, miR-17, miR-92a-1, miR-18a, miR-19a and
miR-19b-1. Of these, three, miR-20a, miR-17 and miR-92a-1, target
six genes, chemoikine (C-X-C motif) ligand 8 (CXCL8),
mitogen-activated protein kinase (MAPK) 9, TP63,
retinoblastoma (RB) 1, RUNX1 and BCL2. The six miRs
are regulated by signal transducer and activator of transcription
5B (STAT5B). miR-19a, miR-18a and miR-19b-1 do not target
any dysregulated genes. In the present study, 10 dysregulated genes
are host genes, however, their miRs are not associated with
dysregulation in CML, for example, RUNX1 includes miR-802
that is not associated with dysregulation in CML. Although certain
host genes are not associated with dysregulation they may be
involved in CML.
Associated network of CML
There are numerous genes and miRs in the
CML-associated network, as presented in Fig. 2, which demonstrates a portion of
the associated network in CML. Similarities and differences between
the dysregulatory network and the associated network could not be
clearly observed, thus a subnetwork was used to examine the two
networks. As presented in Fig. 3,
the larger nodes represent dysregulated data (genes and miRs),
while the smaller nodes represent non-dysregulated data (genes and
miRs). Fig. 3 demonstrates the
associated network including additional TFs, miRs, target genes and
their additional pathways, such as the TP53→miR-29a→protein
phosphatase, Mg2+/Mn2+ dependent, 1D (PPM1D) pathway.
TP53 is a dysregulated gene in CML, while miR-29a and
PPM1D are non-dysregulated. In another pathway, Spi-1
proto-oncogene (SPI1)→miR-155→Fli-1 proto-oncogene, ETS
transcription factor (FLI1), SPI1 and miR-155 have
dysregulatory expression, while FLI1 is a non-dysregulated
gene in CML. The associated network extends the dysregulatory
network and these novel pathways may contribute to tumor growth,
migration, development, or prevention, diagnosis and other
processes in CML.
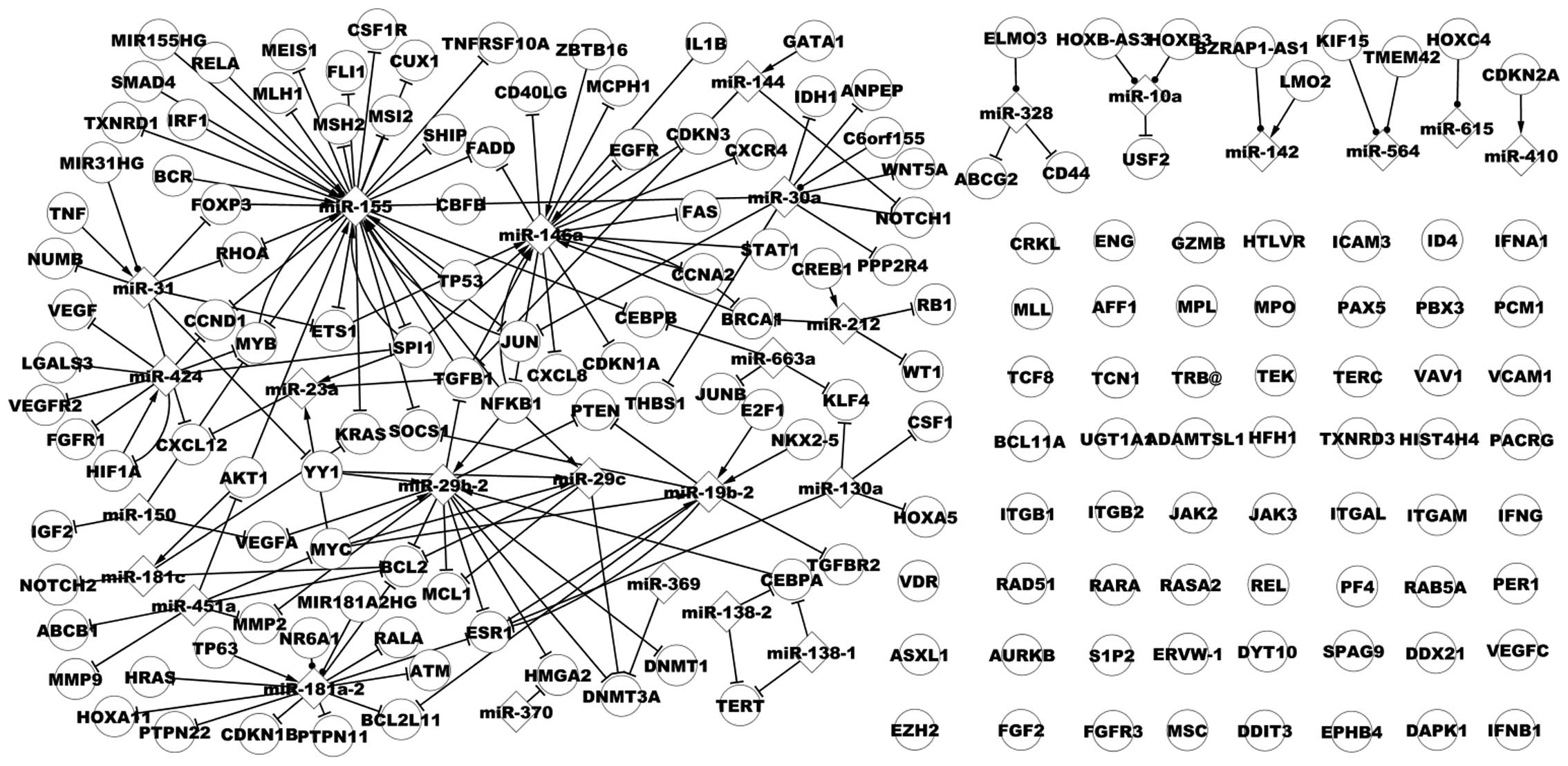 | Figure 2A subnet of associated network in
chronic myelogenous leukemia. miR, microRNA; RELA, v-rel
avian reticuloendotheliosis viral oncogene homolog A; FLI1,
Fli-1 proto-oncogene, ETS transcription factor; IRF1,
interferon regulatory factor 1; EGFR, epidermal growth
factor receptor; TP53, tumor protein 53; MYB, v-myb
avian myeloblastosis viral oncogene homolog; SPI1, Spi-1
proto-oncogene; RB1, retinoblastoma 1; PAX5, paired
box 5; IL8, interleukin-8; NFKB1, nuclear factor of
kappa light polypeptide gene enhancer in B-cells 1; E2F1,
E2F transcription factor 1; KRAS, Kirsten rat sarcoma viral
oncogene homolog; NKX2-5, NK2 homeobox 5; BCL2,
B-cell CLL/lymphoma 2; TP63, tumor protein p63;
BCL2L11, BCL2-like 11. |
 | Figure 3Similarities and differences between
dysregulatory network and associated network in chronic myelogenous
leukemia. miR, microRNA; MYB, v-myb avian myeloblastosis
viral oncogene homolog; FLI1, Fli-1 proto-oncogene, ETS
transcription factor; KRAS, Kirsten rat sarcoma viral
oncogene homolog; SPI1, Spi-1 proto-oncogene; NFKB1,
nuclear factor of kappa light polypeptide gene enhancer in B-cells
1; IL8, interleukin-8; BCL2, B-cell CLL/lymphoma 2;
RB1, retinoblastoma 1; TP53, tumor protein 53;
PPM1D, protein phosphatase, Mg2+/Mn2+ dependent, 1D. |
Transcriptional network of predicted
TFs
Analysis of 11 dysregulated miRs that are regulated
by predicted TFs was conducted. Fig.
4 presents six predicted TFs that regulate nine dysregulated
miRs, which, in turn, target nine dysregulated target genes in CML.
Interferon regulatory factor 1 (IRF1) (33), paired box 5 (PAX5) (2), RUNX1 (34) and nuclear factor of kappa light
polypeptide gene enhancer in B-cells 1 (NFKB1) (35) have been experimentally validated in
CML. The present study focuses on NFKB1, which regulates
four miRs, miR-21, miR-17, miR-146a and miR-155. NFKB1
regulates miR-17, which targets CXCL8, RUNX1,
BCL2, retinoblastoma 1 (RB1) and mitogen-activated
kinase 9 (MAPK9). E2F transcription factor 1 and NK2
homeobox 5 (NKX2-5) co-regulate miR-18a, which targets
Kirsten rat sarcoma viral oncogene homolog (KRAS). In
addition, Fig. 4 demonstrates that
a dysregulated miR may be regulated by various TFs, a target gene
may be targeted by numerous dysregulated miRs and a TF may
indirectly affect other genes via certain dysregulated miRs, such
as miR-155, which is regulated by v-rel avian reticuloendotheliosis
viral oncogene homolog A (RELA) encoding p65, NFKB1
and IRF1. Three miRs, miR-451a, miR-20a and miR-17 target
BCL2. NKX2-5 regulates miR-17 and miR-20a, which
target RUNX1.
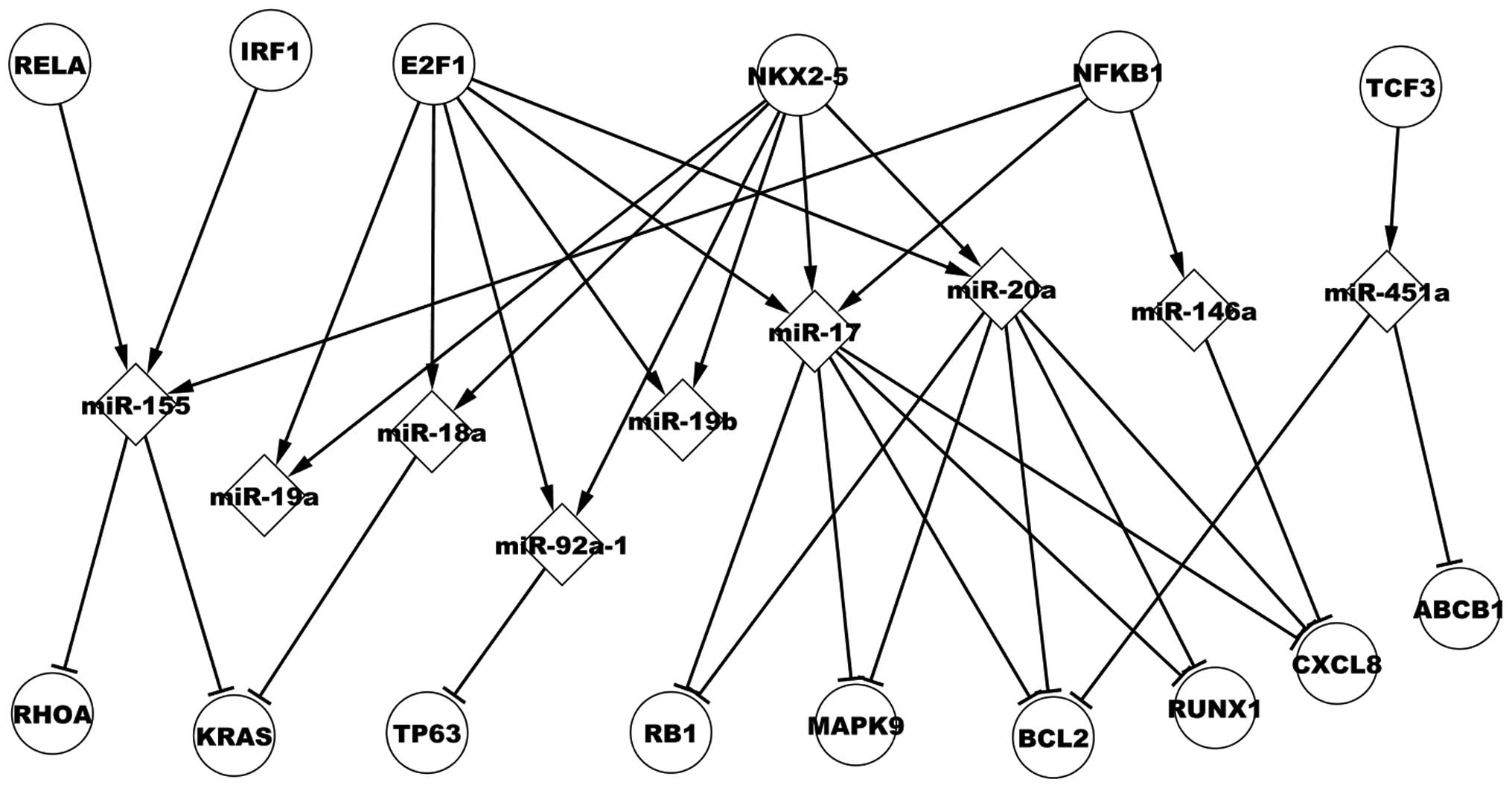 | Figure 4Regulatory associations of predicted
transcriptional network in chronic myelogenous leukemia.
RELA, v-rel avian reticuloendotheliosis viral oncogene
homolog A; IRF1, interferon regulatory factor 1;
E2F1, E2F transcription factor 1; NKX2-5, NK2
homeobox 5; KRAS, Kirsten rat sarcoma viral oncogene
homolog; TP63, tumor protein 63; RB1, retinoblastoma
1; MAPK9, mitogen-activated protein kinase 9; BCL2,
B-cell CLL/lymphoma 2; RUNX1, runt-related transcription
factor 1; IL8, interleukin-8. |
Regulatory pathways of dysregulated
genes
To improve understanding of regulatory pathways,
regulatory pathways of dysregulated genes and miRs, and predicted
TFs were extracted and compared according to their predecessors (a
node preceding the current one in a pathway) and successors (a node
following the current one in a pathway).
V-myb avian myeloblastosis viral oncogene homolog
(MYB) served as an example of a dysregulated gene. Table I indicates MYB, its
predecessors and successors in the three networks. Three miRs
target MYB, which regulates miR-155 in the dysregulatory
network and the association network. Ten miRs target MYB,
which regulates three miRs in the experimentally validated network.
miR-155 and MYB form a feedback loop (FBL) module in the
dysregulatory network. The FBL module is a specific pathway, in
which a TF regulates an miR that targets the TF.
 | Table IRegulatory associations between miRs
and MYB. |
Table I
Regulatory associations between miRs
and MYB.
| Network | MYB
targeting miRs | MYB
regulated miRs |
|---|
| Dysregulatory | miR-150, mir-155,
mir-424 | miR-155 |
| Associated | miR-150, mir-155,
mir-424 | miR-155 |
| Validated | mir-107, miR-150,
miR-155, miR-15a, miR-16-1, miR-16-2, miR-34a, miR-34b, miR-34c,
miR-424 | miR-15a, miR-155,
miR-148a |
Regulatory pathways of dysregulated
miRs
The pathways of each dysregulated miR were extracted
and compared using the same method as for the dysregulated genes.
Table II presents miR-20a, and
its predecessors and successors in the three networks.
STAT5B regulates miR-20a that targets four genes in the
dysregulatory network. Seven TFs regulate miR-20a that targets 15
genes in the associated network. There are 10 TFs, which regulate
miR-20a that targets 26 genes in the experimentally validated
network.
| Table IIRegulatory associations between
miR-20a and genes. |
Table II
Regulatory associations between
miR-20a and genes.
| Network | miR-20a regulating
genes | miR-20a targeted
genes |
|---|
| Dysregulatory | STAT5B | BCL2, RUNX1,
MAPK9, RB1 |
| Associated | CCND1, E2F1,
ESR1, MYC, NKX2-5, SPI1, STAT5B | CCND1, BCL2,
RUNX1, HIF1A, CDKN1A, E2F1, E2F3, MYC, NRAS, MAPK9, PTEN, RB1,
TGFBR2, THBS1, VEGFA |
| Validated | CCND1, E2F1,
MYC, MYCN, NKX2-5, TLX1, TLX3, ESR1, STAT5B, SPI1 | CDKN1A, E2F1,
MUC17, E2F3, HIF1A, MYC, SMAD4, APP, MEF2D, RB1, NRAS, PTEN, MAPK9,
RBL1, RBL2, CCND1, BMPR2, BNIP2, TGFBR2, WEE1, THBS1, VEGFA,
MAP3K12, BCL2, RUNX1, CCND2 |
Regulatory pathways of predicted TFs
The same method was used to extract and compare the
pathways of each predicted TF. E2F1 and NFKB1, as
well as three dysregulated miRs, form three FBL modules. Notably,
IRF1 and RUNX1 are dysregulated genes in CML whereas
NFKB1 and PAX5 are associated with CML.
Table III
presents NFKB1, and its predecessors and successors.
Dysregulated miR (miR-146a) targets NFKB1, which, in turn,
regulates four dysregulated miRs. miR-146a targets NFKB1,
which, in turn, regulates four miRs in the associated network. Nine
miRs target NFKB1, which regulates 24 miRs in the
experimentally validated network. miR-146a and NFKB1 form an
FBL module; NFKB1 is an associated gene in CML and miR-146a
is a dysregulated miR.
| Table IIIRegulatory associations between miRs
and NFKB1. |
Table III
Regulatory associations between miRs
and NFKB1.
| Network | NFKB1
targeting miRs | NFKB1
regulated miRs |
|---|
Dysregulatory
Associated
Validated | miR-146a
miR-146a
let-7a-1, let-7a-2, let-7a-3, miR-146a, miR-146b, miR-15a miR-9-1,
miR-9-2, miR-9-3 | miR-146a, miR-155,
miR-17, miR-21 miR-146a, miR-155, miR-17, miR-21 miR-16-1,
miR-16-2, miR-199a-2, miR-21, miR-214, miR-224, miR-29a, miR-29b-1,
miR-29b-2, miR-29c, miR-34a, miR-365, miR-448, miR-9-1, miR-9-2,
miR-9-3 |
Discussion
In the present study, the regulatory network
consisted of dysregulated genes and miRs, termed a CML dysregulated
expression network. In this network, certain dysregulated genes and
miRs are involved in the occurrence of CML (5,6).
Therefore, these dysregulated genes and miRs as well as their
associations may contribute to the pathogenesis of CML.
In the current study, significant pathways of
dysregulated genes and miRs were observed in CML, such as
miR-17→BCL2, IRF1→miR-155 and
miR-150→MYB→miR-155. These pathways may exert key biological
functions in CML and may be involved in the development of CML.
Certain pathways have been observed to influence processes in CML,
for example, miR-150→MYB contributes to Bcr-Abl-mediated
transformation in CML (25).
Certain pathways have not been observed in CML, however, as they
influence specific processes of other types of cancer, their
biological functions may contribute to CML. For example,
miR-19a→BCL2-like 11 in T cell acute lymphoblastic leukemia
(36) or epidermal growth factor
receptor→miR-21 in age- and mutagen-associated changes in colon
cancer stem-like cells (37). The
remaining pathways that have not been identified in cancers may
function in CML, for example IRF1→miR-155. For the pathways
of predicted TFs, certain pathways have been determined in other
types of carcinoma, for example NKX2-5→miR-17-92 where the
TFs concomitantly reduce E2F1, thereby enhancing survival of
leukemic T cells (38).
In the dysregulated network, genes and miRs exhibit
dysregulated expression. If this dysregulated expression pattern
was to revert to a normal expression pattern, CML may not occur.
Regulation of the dysregulated network may be an efficient gene
therapy strategy for the treatment of CML. Future studies will
focus on the signaling pathways composed of dysregulated genes and
miRs, and explore how these pathways function in CML.
Acknowledgments
The present study was supported by the Jilin
Sino-Rok Institute of Animal Science, Changchun Science and
Technology (grant no. 14KG066), and the Jilin Provincial Science
and Technology Department (grant no. 20130302012NY).
Abbreviations:
|
miR
|
microRNA
|
|
TFs
|
transcription factors
|
|
CML
|
chronic myelogenous leukemia
|
|
NCBI
|
National Center for Biotechnology
Information
|
|
TFBSs
|
transcription factor binding sites
|
|
SNP
|
single nucleotide polymorphism
|
References
|
1
|
Vardiman JW: The World Health Organization
(WHO) classification of tumors of the hematopoietic and lymphoid
tissues: An overview with emphasis on the myeloid neoplasms. Chem
Biol Interact. 184:16–20. 2010. View Article : Google Scholar
|
|
2
|
Kęsy J and Januszkiewicz-Lewandowska D:
Genes and childhood leukemia. Postepy Hig Med Dosw (Online).
69:302–308. 2015. View Article : Google Scholar
|
|
3
|
Gao S, Bajrami I, Verrill C, Kigozi A,
Ouaret D, Aleksic T, Asher R, Han C, Allen P, Bailey D, et al: Dsh
homolog DVL3 mediates resistance to IGFIR inhibition by regulating
IGF-RAS signaling. Cancer Res. 74:5866–5877. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rokah OH, Granot G, Ovcharenko A, Modai S,
Pasmanik-Chor M, Toren A, Shomron N and Shpilberg O: Downregulation
of miR-31, miR-155, and miR-564 in chronic myeloid leukemia cells.
PLoS One. 7:e355012012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamaguchi H, Inokuchi K, Sakuma Y and Dan
K: Mutation of the p51/p63 gene is associated with blastic crisis
in chronic myelogenous leukemia. Leukemia. 15:1729–1734. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Agirre X, Jiménez-Velasco A, San
José-Enériz E, Garate L, Bandrés E, Cordeu L, Aparicio O, Saez B,
Navarro G, Vilas-Zornoza A, et al: Down-regulation of hsa-miR-10a
in chronic myeloid leukemia CD34+ cells increases USF2-mediated
cell growth. Mol Cancer Res. 6:1830–1840. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miething C, Grundler R, Mugler C, Brero S,
Hoepfl J, Geigl J, Speicher MR, Ottmann O, Peschel C and Duyster J:
Retroviral insertional mutagenesis identifies RUNX genes involved
in chronic myeloid leukemia disease persistence under imatinib
treatment. Proc Natl Acad Sci USA. 104:4594–4599. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Y, Yang L, Zhao M, Zhu S, Kang R,
Vernon P, Tang D and Cao L: Targeting microRNA-30a-mediated
autophagy enhances imatinib activity against human chronic myeloid
leukemia cells. Leukemia. 26:1752–1760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hobert O: Gene regulation by transcription
factors and microRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Latchman DS: Transcription factors: An
overview. Int J Biochem Cell Biol. 29:1305–1312. 1997. View Article : Google Scholar
|
|
11
|
Chen K and Rajewsky N: The evolution of
gene regulation by transcription factors and microRNAs. Nat Rev
Genet. 8:93–103. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Betel D, Wilson M, Gabow A, Marks DS and
Sander C: The http://microRNA.orgurisimplemicroRNA.org resource:
Targets and expression. Nucleic Acids Res. 36(Database): D149–D153.
2008. View Article : Google Scholar
|
|
13
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krek A, Grün D, Poy MN, Wolf R, Rosenberg
L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M
and Rajewsky J: Combinatorial microRNA target predictions. Nat
Genet. 37:495–500. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Papadopoulos GL, Reczko M, Simossis VA,
Sethupathy P and Hatzigeorgiou AG: The database of experimentally
supported targets: A functional update of TarBase. Nucleic Acids
Res. 37(Database): D155–D158. 2009. View Article : Google Scholar :
|
|
16
|
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: miRTarBase: A
database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res. 39(Database): D163–D169. 2011.
View Article : Google Scholar
|
|
17
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37(Database): D105–D110. 2009.
View Article : Google Scholar
|
|
18
|
Rodriguez A, Griffiths-Jones S, Ashurst JL
and Bradley A: Identification of mammalian microRNA host genes and
transcription units. Genome Res. 14(10A): 1902–1910. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baskerville S and Bartel DP: Microarray
profiling of microRNAs reveals frequent coexpression with
neighboring miRs and host genes. RNA. 11:241–247. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao G, Huang B, Liu Z, Zhang J, Xu H, Xia
W, Li J, Li S, Chen L, Ding H, et al: Intronic miR-301 feedback
regulates its host gene, ska2, in A549 cells by targeting MEOX2 to
affect ERK/CREB pathways. Biochem Biophy Res Commun. 396:978–982.
2010. View Article : Google Scholar
|
|
21
|
Vaidya S, Ghosh K, Shanmukhaiah C and
Vundinti BR: Genetic variations of hOCT1 gene and CYP3A4/A5 genes
and their association with imatinib response in Chronic Myeloid
Leukemia. Eur J Pharmacol. 765:124–130. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fallah P, Amirizadeh N, Poopak B, Toogeh
G, Arefian E, Kohram F, Hosseini Rad SM, Kohram M, Teimori Naghadeh
H and Soleimani M: Expression pattern of key microRNAs in patients
with newly diagnosed chronic myeloid leukemia in chronic phase. Int
J Lab Hematol. 37:560–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang J, Lu M, Qiu C and Cui Q: TransmiR: A
transcription factor-microRNA regulation database. Nucleic Acids
Res. 38(Database): D119–D122. 2010. View Article : Google Scholar
|
|
24
|
Chekmenev DS, Haid C and Kel AE: P-Match:
transcription factor binding site search by combining patterns and
weight matrices. Nucleic Acids Res. 33(Web Server): W432–W437.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Flamant S, Ritchie W, Guilhot J, Holst J,
Bonnet ML, Chomel JC, Guilhot F, Turhan AG and Rasko JE: Micro-RNA
response to imatinib mesylate in patients with chronic myeloid
leukemia. Haematologica. 95:1325–1333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
27
|
Dong W, Zhang J, Shao N, Tian T, Li L,
Jian J, Zang S, Ma D and Ji C: Development and immunological
evaluation of HLA-specific chronic myeloid leukemia polyepitope
vaccine in Chinese population. Vaccine. 32:3501–3508. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sekiguchi Y, Shimada A, Matsuzawa M, Imai
H, Wakabayashi M, Sugimoto K, Nakamura N, Sawada T, Arita J,
Komatsu N and Noguchi M: Occurrence of carcinoma of the pancreas
following nilotinib therapy for chronic myeloid leukemia: Report of
a case with review of the literature. Turk J Haematol. 32:257–262.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Safran M, Dalah I, Alexander J, Rosen N,
Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T and Krug H:
GeneCards version 3: The human gene integrator. Database (Oxford).
baq0202010.
|
|
30
|
Fujita PA, Rhead B, Zweig AS, Hinrichs AS,
Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H and Coelho
A: The UCSC Genome Browser database: Update 2011. Nucleic Acids
Res. 39(Database): D876–D882. 2011. View Article : Google Scholar :
|
|
31
|
Matys V, Fricke E, Geffers R, Gössling E,
Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis
OV, et al: TRANSFAC: Transcriptional regulation, from patterns to
profiles. Nucleic Acids Res. 31:374–378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T
and Cui Q: HMDD v2.0: A database for experimentally supported human
microRNA and disease associations. Nucleic Acids Res. 42(D1):
D1070–D1074. 2014. View Article : Google Scholar :
|
|
33
|
Tzoanopoulos D, Speletas M, Arvanitidis K,
Veiopoulou C, Kyriaki S, Thyphronitis G, Sideras P, Kartalis G and
Ritis K: Low expression of interferon regulatory factor-1 and
identification of novel exons skipping in patients with chronic
myeloid leukaemia. Br J Haematol. 119:46–53. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao LJ, Wang YY, Li G, Ma LY, Xiong SM,
Weng XQ, Zhang WN, Wu B, Chen Z and Chen SJ: Functional features of
RUNX1 mutants in acute transformation of chronic myeloid leukemia
and their contribution to inducing murine full-blown leukemia.
Blood. 119:2873–2882. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reuter S, Charlet J, Juncker T, Teiten MH,
Dicato M and Diederich M: Effect of curcumin on nuclear factor
kappaB signaling pathways in human chronic myelogenous K562
leukemia cells. Ann N Y Acad Sci. 1171:436–447. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J,
Yuan P, Zhong Z, Li Q, Jia H, et al: MicroRNA and transcription
factor co-regulatory network analysis reveals miR-19 inhibits CYLD
in T-cell acute lymphoblastic leukemia. Nucleic Acids Res.
40:5201–5214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nautiyal J, Du J, Yu Y, Kanwar SS, Levi E
and Majumdar AP: EGFR regulation of colon cancer stem-like cells
during aging and in response to the colonic carcinogen
dimethylhydrazine. Am J Physiol Gastrointest Liver Physiol.
302:G655–G663. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nagel S, Venturini L, Przybylski GK,
Grabarczyk P, Schmidt CA, Meyer C, Drexler HG, Macleod RA and
Scherr M: Activation of miR-17–92 by NK-like homeodomain proteins
suppresses apoptosis via reduction of E2F1 in T-cell acute
lymphoblastic leukemia. Leuk Lymphoma. 50:101–108. 2009. View Article : Google Scholar : PubMed/NCBI
|














