Introduction
Hepatocellular carcinoma (HCC) is the sixth most
frequently diagnosed type of cancer and the third leading cause of
cancer mortality (1). In Asia,
liver cancer is one of the most common types of cancer (1). The majority of cases are associated
with chronic liver disease, including cirrhosis or hepatitis
(2). The development of HCC
involves sequential morphological changes from hepatic tissue with
no recorded tumor complication, to preneoplastic lesions and
subsequently to neoplastic lesions (3,4). HCC
can be removed through surgical liver resection and liver
transplantation. However, the survival rate of patients who have
already had surgical resection is <25% (5) and liver cancer often reoccurs within
2 years. Furthermore, current chemotherapeutic drugs cannot
effectively control tumor progression, and drug resistance is
common. Therefore, novel chemotherapeutic agents are urgently
required to inhibit HCC at an early stage, to improve the overall
survival rate of patients with HCC.
Several rodent HCC models have been developed to
investigate the pathogenesis, treatment and prevention of liver
cancer. Chemically-induced hepatocarcinogenesis provides a valuable
model for investigating the molecular biology of
hepatocarcinogenesis, particularly in its early stages, for various
reasons cited previously (6).
Diethylnitrosamine (DEN), a DNA alkylating agent, is a widely used
to induce carcinogenesis in the rodent model of HCC.
Metformin, a biguanide, is one of the most widely
prescribed oral glucose-lowering agents used in the treatment of
type II diabetes (7) and has a
very low rate of toxicity. In addition to its antidiabetic effects,
various in vitro and in vivo experiments revealed the
anticancer characteristics of metformin (8–11).
Previously, other groups revealed that metformin effectively
inhibits HCC in mice (12). In
addition, the efficiency of metformin in decreasing the incidence
of liver cancer has been supported by other in vitro,
epidemiological and animal studies (13,14),
supporting the potential of its use as an anticancer drug.
In addition, inactivation of AMP-activated protein
kinase (AMPK) is associated with tumorigenesis and several
malignant cancer types. Zheng et al (15) observed the phosphorylation status
of AMPK in HCC and revealed that AMPK acts as a negative regulator
in the liver, and loss of the inhibitory effects of AMPK
contributes to the progression and invasion of HCC (15). The effect of metformin on HCC cells
is predominantly dependent on the activation of AMPK (15). However, whether certain of the
antitumor effects of metformin on HCC in vivo are dependent
of AMPK remains to be elucidated.
In the present study, the effects of metformin on
the development and recurrence of HCC were investigated using the
DEN-induced rat HCC model. To investigate whether metformin affects
tumor growth in vivo, a long-term DEN-induced animal model
of HCC, causing a high incidence of malignant tumor generation and
cirrhosis similarly observed in humans, was used. Concomitant
treatment with DEN and metformin was performed to determine whether
any metformin-mediated inhibitory effects occurred during the
development of HCC.
Materials and methods
Chemicals and reagents
DEN was purchased from Sigma-Aldrich (St. Louis, MO,
USA). Metformin hydrochloride was supplied by Danashmand Organic
Private Ltd. (Maharashtra, India), and its purity was determined
using standard and high-performance liquid chromatography.
Metformin was dissolved in water at a concentration of 0.5 mg/ml
and DEN was dissolved in 0.9% saline at 50 mg/kg. All antibodies
were purchased from Abcam (Cambridge, MA, USA), unless otherwise
stated. All molecular biology reagents were molecular biology grade
or higher, unless otherwise stated.
Animals, husbandry, and experimental
design
A total of 200 male, 6-week-old, Wistar rats, with a
mean body weight of 180.75±5.5 g, were obtained from Orient Bio,
Inc. (Yongin, Kyungki, Korea) and were quarantined for 7 days prior
to the initiation of the investigation. The rats were maintained at
the laboratory animal facility of the Asan Institute for Life
Sciences under specific pathogen-free conditions, as indicated by
the guidelines of the Animal Care and Use Committee of Asan
Institute for Life Sciences.
The rats were housed in environmental conditions
with a temperature of 20–24°C, relative humidity of 50±10%, 12 h
light/dark cycles, illumination at 150–300 Lux and ventilation
10–20 times/hour, which were monitored every hour for 24 h and
maintained within acceptable ranges throughout the present study.
The rats were housed three per cage at the beginning of the study
and fed an autoclaved pellet diet (Lab Diet #5002; PMI Nutrition
International LLC, St. Louis, MO, USA) ad libitum.
The study protocol was reviewed and approved by the
Institutional Animal Care and Use Committee of Asan Institute for
Life Sciences (IACUC no. 2012-01-208). The rats were divided into
four groups (60 rats/group in A and B; 40 rats/group in C and D).
All rats in Groups A and B received weekly intraperitoneal
injections of 50 mg/kg body weight of DEN dissolved in 0.9% NaCl
solution (Daehan Pharm Co, Ltd., Kyungki, Korea) over a period of
16 weeks. The rats in groups B and C received ad libitum
access to 0.5 mg/ml metformin dissolved in the drinking water.
Group D, the negative control group, was provided drinking water
ad libitum. All clinical findings were recorded daily, and
body weights, food and water intake were measured on a weekly
basis. At the end of the study, all animals were weighed and
anesthetized with 4% isoflurane (cat. no. 400-326-09; Baxter,
Deerfield, IL, USA). The animals were subsequently sacrificed by
exsanguination from the abdominal aorta.
Histopathological and immunohistochemical
analysis
On completion of the treatment, the rats were
euthanized under isoflurane anesthesia, and their livers were
excised and weighed by electronic microbalance (Ohaus Corporation,
Pine Brook, NJ, USA). Following a hepatectomy in all rats, the
number of surface nodules on the liver was grossly investigated by
visual inspection [Miss Woori Jo (University of Ulsan College of
Medicine) performed the gross inspection in a blinded manner].
For histopathological and immunohistochemical
evaluation, and western blot analysis, the medial, left and right
lateral lobes of the liver and the tumor lesions were separated,
fresh-frozen at −196°C in liquid nitrogen and stored at −80°C until
further analysis. The remainder of the liver tissue was fixed in
10% neutral buffered formalin (Sigma-Aldrich) for histological
evaluation. A tissue processor (Thermo Fisher Scientific, Inc.,
Runcorn, UK) was used to prepare liver samples from the
formalin-fixed samples for analysis by fixing, staining and
dehydrating. The paraffin-embedded tissue blocks were cut at a
3-µm thickness and mounted onto glass slides. Staining was
performed with hematoxylin (YD-Diagnostics, Kyungki, Korea) and
eosin (BBC Biochemicals, Mount Vernon, WA, USA) using an
autostainer (Leica ST5015; Leica Biosystems Nussloch GmbH,
Nussloch, Germany). Immunohistochemistry was performed using an
automated slide preparation system, Benchmark XT (Ventana Medical
Systems, Inc., Tucson, AZ, USA). Deparaffinization, epitope
retrieval and immunostaining were performed, according to the
manufacturer's instructions, using cell conditioning solutions and
the Benchmark ultra-VIEW diaminobenzidine detection system (Ventana
Medical Systems, Inc.). Tumor sections were stained with a
monoclonal rabbit anti-rat anti-glutathione S-transferase placental
form (GST-P) antibody (cat. no. ab138491; 1:2,000 dilution), a
monoclonal, rabbit anti-rat anti-proliferating cell nuclear antigen
(PCNA) antibody (cat. no. ab92552; 1:1,000) and a polyclonal,
rabbit anti-rat anti-cytokeratin 8 (CK8) antibody (cat. no.
ab59400; 1:100). Positive signals were amplified using ultraVIEW
copper (Ventana Medical Systems, Inc.), and the sections were
counterstained with hematoxylin and bluing reagent (Ventana Medical
Systems, Inc.). The number and area of GST-P-positive foci >0.2
mm2, and the total area of each liver section were
measured using a Panoramic 250 Flash II slide scanner (3D-Histech,
Ltd., Budapest, Hungary) and Panoramic viewer software 1.15.2
(3D-Histech, Ltd.) to provide values per cm2 of liver
tissue. PCNA staining was quantified in five fields of 2
mm2 each in six rats from each group. In addition,
staining for the hepatic progenitor cell marker, cytokeratin 8
(CK8), was quantified in an area of 6 mm2 in three rats
from each group. The percentage of the area occupied by the
positive cells in the total area occupied by all cells was analyzed
using i-Solution Lite version 10.6 (IMT i-Solution, Inc.,
Vancouver, BC, Canada) and the results were expressed as the
percentage of positive cell staining.
Western blotting
The liver tissues were homogenized and the samples
were incubated on ice with frequent vortexing for 15 min and
centrifuged for 20 min at 18,000 × g and 4°C. The protein content
of each sample was quantified using a bicinchoninic acid kit
(Pierce, Rockford, IL, USA). Equivalent quantities of protein
lysates (20 µg/lane) were separated on 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis gels [dH2O,
30% acrylamide mix (Bio-Rad Laboratories, Inc., Hercules, CA, USA),
1.5M Tris pH 8.8 (Bio-Rad Laboratories, Inc.), 10% SDS (GenDepot,
Barker, TX, USA), 10% ammonium persulfate (Bio-Rad Laboratories,
Inc.) and PlusOne TEMED (GE Healthcare Life Sciences, Freiburg,
Germany)]. The proteins were transferred onto polyvinylidene
membranes (Bio-Rad Laboratories, Inc.) and the blots were incubated
with 5% non-fat dry milk for 1 h at room temperature, followed by
incubation with the appropriate primary antibodies, including
monoclonal rabbit anti-rat anti-phospho (p−)AMPKα antibody (Cell
Signaling Technology, Inc., Danvers, MA, USA; cat. no. 2535;
1:500), polyclonal rabbit anti-rat anti-AMPKα antibody (cat. no.
ab131512; 1:500) for 20 h at 4°C. The blots were washed with 0.1%
Tris-buffered saline, containing 1% Tween-20, prior to incubation
with anti-rabbit (1:10,000) immunoglobulin G peroxidase-conjugated
secondary antibodies (cat. no. SA002-500; GenDepot), for 1 hour at
room temperature. The membranes were developed with
chemiluminescent substrates (Thermo Fisher Scientific, Inc.).
Hybridization with monoclonal mouse anti-rat anti-β-actin antibody
(cat. no. ab8224; 1:4,000) was used to confirm equal protein
loading.
Statistical analysis
Statistical significance was determined using
GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). The data
are presented as the mean ± standard deviation, with the number of
samples indicated in the figure legend. The statistical
significance of the differences between groups was analyzed by
Student's t-test and a two-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
Survival rate, tumor-free state and body
weight of rats following treatment
The experimental design is shown in Fig. 1A, where the rats in groups B and C
received 0.5 mg/ml metformin dissolved in drinking water and rats
in groups A and B were treated with 50 mg/kg DEN by intraperitoneal
injections weekly for 16 weeks. The survival rate, mean body weight
and tumor-free state were determined for each of the groups
(Fig. 1), A total of three rats in
Group A died during the duration of the experiment. The deaths
occurred on days 87, 101 and 115. The survival rate is shown in
Fig. 1C. Fig. 1D shows the tumor-free state
throughout the present study. Additionally, the mean body weight
during the duration of the investigation is shown in Fig. 1B. The body weights of the rats in
Groups A and B were 17% lower compared with those in Groups C and D
by the 10th week. Groups A and B, which were DEN-treated
groups, demonstrated a significant reduction in body weight
compared with the control group from the second week. However, no
difference was observed between group A and B. The loss of body
weight in the DEN-treated groups, A and B, and the deaths of rats
in Group A, were likely due to the general toxic effects of DEN.
Since no deaths occurred in Group B, it is possible that metformin
protected the rats from the severe toxicity caused by DEN.
Gross examination and histopathological
analysis
Fig. 2A shows the
gross picture and the number of liver surface nodes observed upon
gross examination. The number of liver nodules gradually increased
over time (Fig. 2B).
Representative hematoxylin and eosin micrographs are shown in
Fig. 2C. Altered hepatocellular
foci (AHF) and tumor nodules were examined in hematoxylin and
eosin-stained slides, and were classified as hepatocellular
adenoma, HCC and hemangio-sarcoma, as shown in Table I. Group B, which was treated with
metformin and DEN, exhibited a decreased incidence of AHF,
hepatocellular adenoma and HCC compared with Group A, which was
treated with DEN alone, between weeks 16 and 18. Cirrhosis was
observed in the long-term DEN-treated group.
 | Table IIncidence of histopathological changes
in the livers of Wistar rats. |
Table I
Incidence of histopathological changes
in the livers of Wistar rats.
| Histopathological
change | Treatment duration
|
|---|
12 weeks
| 14 weeks
| 16 weeks
| 18 weeks
|
|---|
| DEN | DEN + MF | DEN | DEN + MF | DEN | DEN + MF | DEN | DEN + MF |
|---|
| Lesions (n) | 15 | 15 | 15 | 15 | 15 | 15 | 12 | 15 |
| Hepatocellular
adenoma | 0 | 0 | 2 | 0 | 5 | 2 | 3 | 5 |
| Hepatocellular
carcinoma | 0 | 0 | 1 | 0 | 1 | 0 | 6 | 5 |
|
Hemangiosarcoma | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| AHF | 2.87 | 2.07 | 9.27 | 6.27 | 9.00 | 5.53 | 14.50 | 12.87 |
| Total (except
AHF) | 0 | 0 | 0.2 | 0 | 0.47 | 0.13 | 0.75 | 0.73 |
Absolute and relative liver weights
The final body weights, absolute liver weights and
relative liver weights (the relative liver weights were obtained by
dividing the absolute liver weights by the body weights) were
recorded following sacrifice at 12, 14, 16 and 18 weeks (Fig. 2D). Necropsies revealed a marked
enlargement of the liver in rats treated with long-term DEN (Groups
A and B), which was consistent with the increase in liver weight.
Treatment with DEN alone (Group A) markedly increased the relative
liver weight by 150.1%, whereas co-treatment with DEN and metformin
(Group B) resulted in an increase in the relative liver weight of
130.4% compared with the control group at 18 weeks. These data
suggested that the enlargement of the liver caused by DEN was
diminished by treatment with metformin.
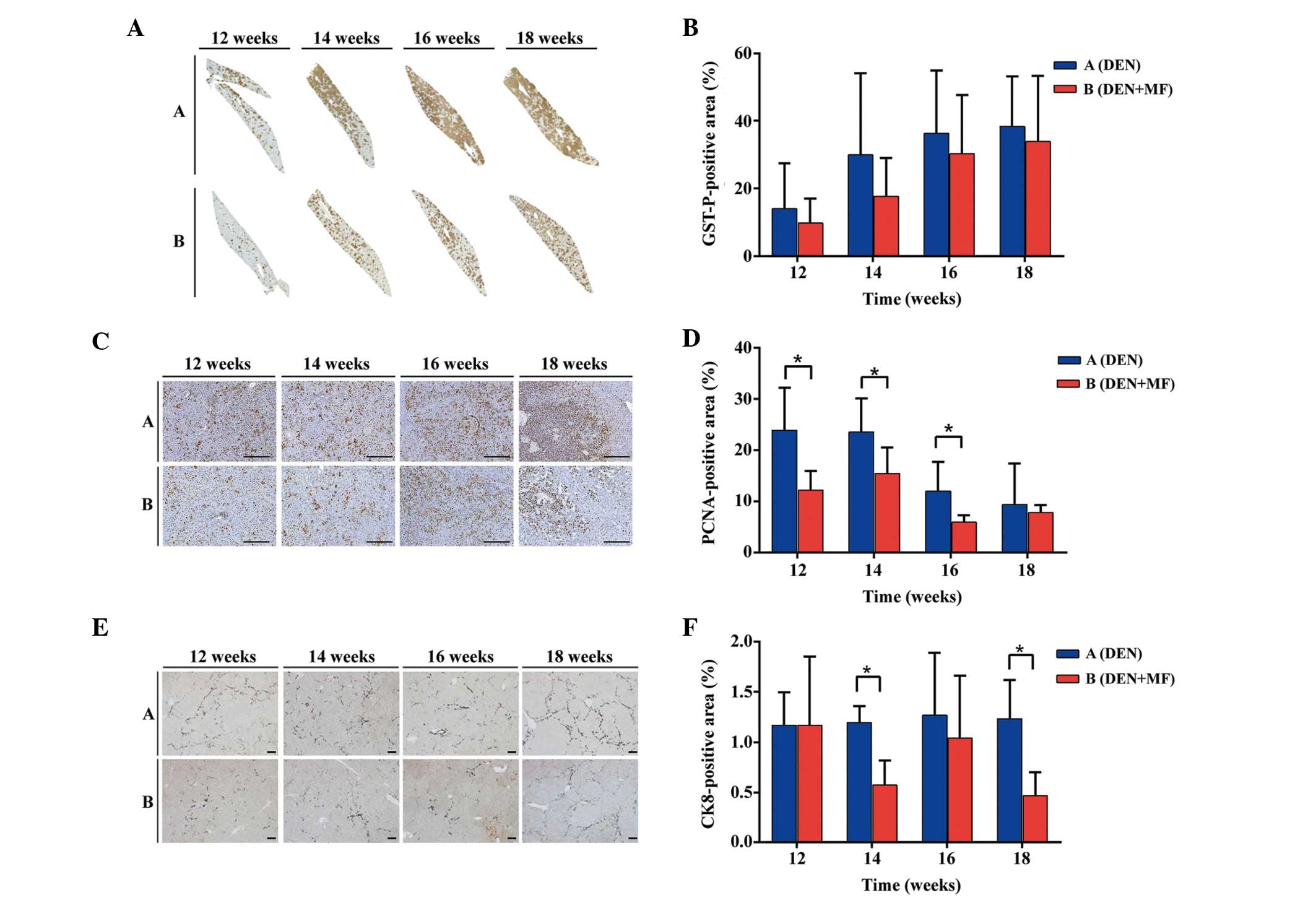
Quantitative evaluation of GST-P positive
cells
The number and area of GST-P-positive cells as
end-point markers for hepatocarcinogenesis, particularly in rats as
opposed to mice (16), were
compared between the DEN-treated rats and the rats co-treated with
metformin. As shown in Fig. 3A and
B, the GST-P-positive cells in each group steadily increased
over time, however, GST-P positivity in Group A remained higher
compared with Group B. These results suggested that a reduction in
GST-P-positive cells may represent one mechanism underlying the
antitumor activity of metformin.
Quantitative evaluation of PCNA-positive
cells
Immunohistochemistry for PCNA was performed to
determine whether metformin acts by inhibiting proliferation, as
previously reported in vitro (17). PCNA-positive cells were quantified
in five high-powered fields in different tumors from
metformin-treated and untreated rats. Differences in PCNA-positive
cells between Groups A and B were observed at weeks 12, 14, 16 and
18 (P<0.05). As shown in Fig. 3C
and D, the difference between Group A and B decreased over
time. This may reflect the formation of tumors from AHF.
Quantitative evaluation of CK8-positive
cells
Hepatic progenitor cells in the rodent liver samples
were assessed by immunohistochemistry for CK8. Hepatic progenitor
cells and reactive ductules were reactive for CK8 (18). The ratio of CK8-positive cells
between Group A and B were 1.001, 1.775, 1.221 and 2.639 at weeks
12, 14, 16 and 18, respectively (P<0.05). Metformin inhibited
the proliferation of hepatic progenitor cells, resulting in reduced
tumor incidence, consistent with other previous in vitro
studies (17). Representative
micrographs are shown in Fig. 3E,
and quantification of CK8 immunostaining for rats in each group is
shown in Fig. 3F.
Western blot analysis
Since metformin is important in AMPK activation, the
activation status of AMPK was examined in the liver tissues in the
HCC model. The total AMPKα and p-AMPKα levels were analyzed in six
rat livers in each group. DEN treatment significantly reduced the
level of p-AMPKα1, however, the levels of p-AMPKα1 were increased
in the co-treatment groups (Fig.
4). These results suggested that the antitumor effect of
metformin is, at least partially, dependent on AMPKα1
activation.
Discussion
In the present study, the number of surface foci,
the relative liver weights, the size of GST-P-positive lesions, and
the percentage of PCNA- and CK8-positive liver sections were
determined to confirm the antitumorigenic effect of metformin on
the liver. The data demonstrated that metformin successfully
inhibited the occurrence of early-stage hepatocarcinogenesis
induced by DEN.
Previous studies performed with animal models
treated with DEN have been relatively short-term exposures of <4
weeks and rarely induced cirrhosis. Cirrhosis is one of numerous
types of liver injury, which can cause HCC. In the present study,
DEN was administered up to 16 weeks, which was relatively longer
compared with other previous studies. The rats in group A and B
received a sufficient exposure to DEN in order to create an animal
model, which resembles human HCC. Furthermore, the majority of HCC
occurs in livers that are chronically injured as a result of liver
cirrhosis or chronic hepatitis. This demonstrates that the
initiation of HCC may occur far before HCC can be clinically
detected. Certain hepatocarcinogen treatment regimens, including
chronic treatment of rats with DEN, also induce AHF, megalocytosis,
cholangiofibrosis, cholangiocarcinoma and liver cirrhosis, which
are preceded by extensive steatosis, extensive fibrosis and nodular
hyperplasia by hepatocytes. Therefore, our long-term DEN treatment
model may be a valuable animal model to mimic human HCC
tumorigenesis and to assess novel therapies in rats.
The mechanism of action of metformin was
investigated in the present study. To assess whether metformin
treatment resulted in the activation of AMPK, liver tumors were
analyzed for p-AMPK. In the present study, western blot analysis of
HCC tissue revealed a significant decrease in the levels of
p-AMPKα1 in the livers of DEN-treated rats, compared with control
rats or those treated with metformin, alone or in combination with
DEN. Therefore, these results provided clear evidence in support of
the AMPK-dependent effects of metformin on HCC.
The inhibitory actions of metformin on tumor growth,
including cellular studies, are possibly explained by several
mechanisms (19–22). Early previous studies have
suggested that metformin may inhibit tumor growth by activating
AMPK, an energy sensor (19,23,24).
Other studies have demonstrated AMPK-independent effects of
metformin on glucose homeostasis and tumor growth in vitro
and in vivo (25,26). These studies still contradict
certain other studies, including the present study, which
demonstrated AMPK activation in the liver by metformin. One
possible explanation for the discrepancies between these studies
may be the extended duration of treatment compared with other
previous studies, which used a short-term treatment (27). In addition, Memmott et al
(22) have recently demonstrated
that AMPK activation by metformin in the liver may depend on the
route of administration and that intraperitoneal, however, not
oral, metformin treatment increased the phosphorylation of AMPK in
the liver. Intraperitoneal administration is known to result in a
higher systemic concentration of metformin compared with oral
administration (22). However,
oral administration is currently being used in clinical trials, and
therefore, the present study selected oral administration for
investigation. The present study revealed that phosphorylation of
AMPK was increased by oral administration, suggesting that
metformin inhibited tumor growth via an AMPK-dependent pathway in
the liver when administered orally.
This is the first study, to the best of our
knowledge, to demonstrate the antitumor effects of metformin on
long-term DEN-treated rat livers. The long-term DEN-treated rat
model of HCC is a good model reflecting the status of human HCC and
the present study has investigated the inhibitory effects of
metformin on HCC. As in a previous study using AMPK as a biomarker
for HCC (15), the therapeutic
effects of metformin were assessed in a patient-derived xenograft
animal model using p-AMPK as a biomarker for HCC in the stage of
drug development. These studies may offer an improved insight into
our understanding of the effects of metformin on the early stage of
HCC development in rats and may allow for the appropriate
application of metformin for the prevention of HCC recurrence
following treatment of primary HCC. Also, the present study
provided a clear rationale for the use of metformin for the cure
for the high-risk patients.
Acknowledgments
The present study was supported by grants from the
Korean Health Technology Research and Development Project, Ministry
of Health and Welfare, Korea (nos. HI06C0868 and HI10C2014).
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
DEN
|
diethylnitrosamine
|
|
MF
|
metformin
|
|
AHF
|
altered hepatocellular foci
|
|
GST-P
|
glutathione S-transferase placental
form
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
CK8
|
cytokeratin 8
|
|
AMPK
|
AMP-activated protein kinase
|
References
|
1
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bosch FX, Ribes J, Cléries R and Díaz M:
Epidemiology of hepatocellular carcinoma. Clin Liver Dis.
9:191–211. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maronpot RR, Montgomery CA Jr, Boorman GA
and McConnell EE: National toxicology program nomenclature for
hepatoproliferative lesions of rats. Toxicol Pathol. 14:263–273.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eustis SL: The sequential development of
cancer: A morphological perspective. Toxicol Lett. 49:267–281.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wild CP and Hall AJ: Primary prevention of
hepatocellular carcinoma in developing countries. Mutat Res.
462:381–393. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ogawa K: Molecular pathology of early
stage chemically induced hepatocarcinogenesis. Pathol Int.
59:605–622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kirpichnikov D, McFarlane SI and Sowers
JR: Metformin: An update. Ann Intern Med. 137:25–33. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aljada A and Mousa SA: Metformin and
neoplasia: Implications and indications. Pharmacol Ther.
133:108–115. 2012. View Article : Google Scholar
|
|
10
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar
|
|
11
|
Algire C, Zakikhani M, Blouin MJ, Shuai JH
and Pollak M: Metformin attenuates the stimulatory effect of a
high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat
Cancer. 15:833–839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhalla K, Hwang BJ, Dewi RE, Twaddel W,
Goloubeva OG, Wong KK, Saxena NK, Biswal S and Girnun GD: Metformin
prevents liver tumorigenesis by inhibiting pathways driving hepatic
lipogenesis. Cancer Prev Res (Phila). 5:544–552. 2012. View Article : Google Scholar
|
|
13
|
Qu Z, Zhang Y, Liao M, Chen Y, Zhao J and
Pan Y: In vitro and in vivo antitumoral action of metformin on
hepatocellular carcinoma. Hepatol Res. 42:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen HP, Shieh JJ, Chang CC, Chen TT, Lin
JT, Wu MS, Lin JH and Wu CY: Metformin decreases hepatocellular
carcinoma risk in a dose-dependent manner: Population-based and in
vitro studies. Gut. 62:606–615. 2013. View Article : Google Scholar
|
|
15
|
Zheng L, Yang W, Wu F, Wang C, Yu L, Tang
L, Qiu B, Li Y, Guo L, Wu M, et al: Prognostic significance of AMPK
activation and therapeutic effects of metformin in hepatocellular
carcinoma. Clin Cancer Res. 19:5372–5380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morimura S, Suzuki T, Hochi S, Yuki A,
Nomura K, Kitagawa T, Nagatsu I, Imagawa M and Muramatsu M:
Trans-activation of glutathione transferase P gene during chemical
hepatocarcinogenesis of the rat. Proc Natl Acad Sci USA.
90:2065–2068. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saito T, Chiba T, Yuki K, Zen Y, Oshima M,
Koide S, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, et al:
Metformin, a diabetes drug, eliminates tumor-initiating
hepatocellular carcinoma cells. PLoS One. 8:e700102013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lowes KN, Croager EJ, Olynyk JK, Abraham
LJ and Yeoh GC: Oval cell-mediated liver regeneration: Role of
cytokines and growth factors. J Gastroenterol Hepatol. 18:4–12.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rocha GZ, Dias MM, Ropelle ER,
Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ and Carvalheira JB:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kourelis TV and Siegel RD: Metformin and
cancer: New applications for an old drug. Med Oncol. 29:1314–1327.
2012. View Article : Google Scholar
|
|
22
|
Memmott RM and Dennis PA: LKB1 and
mammalian target of rapamycin as predictive factors for the
anticancer efficacy of metformin. J Clin Oncol. 27:e226author reply
e227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zakikhani M, Dowling RJ, Sonenberg N and
Pollak MN: The effects of adiponectin and metformin on prostate and
colon neoplasia involve activation of AMP-activated protein kinase.
Cancer Prev Res (Phila). 1:369–375. 2008. View Article : Google Scholar
|
|
25
|
Foretz M, Hébrard S, Leclerc J,
Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F and
Viollet B: Metformin inhibits hepatic gluconeogenesis in mice
independently of the LKB1/AMPK pathway via a decrease in hepatic
energy state. J Clin Invest. 120:2355–2369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kalender A, Selvaraj A, Kim SY, Gulati P,
Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et
al: Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shaw RJ, Lamia KA, Vasquez D, Koo SH,
Bardeesy N, Depinho RA, Montminy M and Cantley LC: The kinase LKB1
mediates glucose homeostasis in liver and therapeutic effects of
metformin. Science. 310:1642–1646. 2005. View Article : Google Scholar : PubMed/NCBI
|