Introduction
Breast cancer, as the most frequently diagnosed type
of cancer in women, has become the primary cause of
cancer-associated mortality in women worldwide (1). It was estimated that ~1,700,000 new
breast cancer cases were diagnosed in 2012, with 6,300,000 cases of
breast carcinoma-associated mortality occurring in the same year,
according to GLOBOCAN 2012 (1).
Furthermore, the incidence and mortality rates of breast cancer
have increased by >20 and 14%, respectively, since the 2008
statistics (2). The sharp rise in
breast cancer incidence is particularly evident in developing
countries (2). These statistics
emphasize the requirement for novel and effective breast
cancer-targeted therapeutic agents.
The occurrence and development of cancer is a
complex process, which ultimately leads to the deregulation of cell
signaling pathways that govern cell proliferation and survival
(3–5). Over the past two decades, there has
been a shift from conventional treatments, including surgery,
radiotherapy and chemotherapy, towards more specific targeted
therapies (6). Breast cancer is a
heterogeneous disease comprising multiple subtypes with different
molecular signatures, prognoses and responses to therapies
(7). As knowledge of the
underlying molecular mechanisms responsible for tumor development
and chemotherapeutic resistance has increased, contemporary
treatment of breast cancer has entered an era of targeted therapies
based on molecular typing, and examples of these molecular targeted
therapeutics for breast cancer include tyrosine kinase inhibitors
(TKIs), angiogenesis inhibitors and agents that perturb DNA repair
(8). In addition, reactivation of
the apoptotic program in order to overcome resistance of tumor
cells to cell death is being pursued as a novel cancer therapeutic
strategy (9,10).
Apoptosis is genetically programmed cell death,
which operates via distinct biochemical and genetic pathways; these
pathways are also utilized during development and homeostasis in
normal tissues (11). Consistent
with this, the evasion of apoptosis is denoted as one of the key
'hallmarks' of cancer (4).
Apoptotic pathways include the mitochondrial/intrinsic pathway and
the death receptor/extrinsic pathway; and each of these signaling
cascades activates caspases, which are the critical effector
molecules of apoptosis. The intrinsic apoptotic pathway is
initiated by permeabilization of the mitochondria outer membrane,
which leads to the activation of caspase 9, whereas the extrinsic
pathway is triggered by ligand binding to death receptors, which
leads to caspase 8 activation. Subsequently, the activation of
effector caspases 3 and/or 7 leads to the cleavage of downstream
substrates and the final execution of apoptosis (12,13).
The upregulation of inhibitor of apoptosis proteins
(IAPs) is a mechanism by which tumor cells evade apoptosis. IAPs
with one or more baculovirus IAP repeat domains belong to a family
of key apoptosis regulators and are overexpressed in multiple human
malignancies (14); thus, they are
relevant targets for therapeutic intervention. In order to execute
apoptosis, the activity of IAPs can be inhibited when they are
bound to second mitochondria-derived activator of caspase
(Smac)/direct IAP-binding protein with low PI (DIABLO), which is
released from mitochondria into the cytosol. X-linked IAP (XIAP),
cIAP1 and cIAP2, which are three major members of the IAPs family,
are all targeted by Smac (15,16).
Another form of programmed cell death, termed
'necroptosis', proceeds via a caspase-independent route. When
either caspase 8 or FLICE-like inhibitory protein are absent, or
the activation or function of caspase 8 or Fas-associated death
domain (FADD) is suppressed, phosphorylated receptor-interacting
protein kinase-1 (RIP1), receptor-interacting kinase-3 (RIP3) and
mixed lineage kinase domain-like protein assemble into a complex
referred to as the 'necrosome' (17–20).
However, the mechanisms mediating the execution of necroptosis
remain to be fully elucidated. This is a critical gap in current
understanding, as necroptosis-associated events are implicated in
the pathophysiological processes of several diseases, including
myocardial infarction and stroke (21,22),
ischemia-reperfusion injury (23,24),
atherosclerosis (25) and other
common clinical disorders. Thus, an improved knowledge of
necroptosis may lead to the identification of necroptosis
inhibitors, and thus provide a novel collection of therapeutic
agents to modulate this alternate form of cell death.
LCL161 is a small, orally available Smac mimetic
compound, which binds to and triggers the degradation of several
IAPs; which is then associated with the induction of apoptosis via
caspase activation (26). LCL161
was designed to mimic the AVPI tetrapeptide binding motif at the
Smac N-terminus, as this region is required for binding to XIAP,
cIAP1 and cIAP2. Upon binding to cIAP1, LCL161 triggers the
autoubiquitination and proteasomal degradation of cIAP1, which is
followed by nuclear factor-κB activation and tumor necrosis
factor-α-dependent apoptosis (27). LCL161 also potentiates the
anti-leukemic effects of TKIs (28).
The present study was designed to evaluate the
effect of LCL161 in human breast cancer cells, and to delineate the
molecular mechanisms by which LCL161 causes cell death.
Materials and methods
Reagents and antibodies
The LCL161 Smac mimetic was purchased from Active
Biochemicals Co., Ltd. (Hong Kong, China) and dissolved in dimethyl
sulfoxide (DMSO; BioSharp, Hefei, China) as a stock solution of 1
mmol·l−1. Rabbit anti-XIAP (cat. no. bs-1281R; 1:500)
and rabbit anti-cIAP1 (cat. no. bs-4262R; 1:500) were purchased
from Bioss, Inc (Hong Kong, China). Rabbit anti-cIAP2 (cat. no.
AP6142a; 1:1,000) was purchased from Abgent, Inc. (Shuzhou, China),
and rabbit anti-B-cell lymphoma 2 (Bcl-2)-associated X protein
(BAX; cat. no. 50599-2-lg; 1:2,000) and rabbit anti-Bcl-2 (cat. no.
12789-1-AP; 1:1,000) were purchased from Proteintech Group, Inc.
(Chicago, IL, USA). Rabbit anti-myeloid cell leukemia (Mcl)-1 (cat.
no. sc-819; 1:500) and rabbit anti-RIP1 (cat. no. sc-7881; 1:500)
were purchased from Santa Cruz Biotechnology, Inc., (Dallas, TX,
USA). Rabbit anti-β-actin (cat. no. BL005A; 1:1,000) was purchased
from BioSharp. An Annexin V Fluorescein isothiocyanate
(FITC)/propidium iodide (PI) Apoptosis Detection kit was purchased
from KeyGen Biotech Co., Ltd. (Nanjing, China).
Cell culture
MDA-MB-231 and MCF-7 human breast cancer cell lines
were obtained from Shanghai Cell Bank, (Shanghai, China). The cells
were cultivated in Dulbecco's modified Eagle's medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 44
mmol·l−1 sodium bicarbonate, 20 Iu·l−1
penicillin, and 20 Iu·l−1 streptomycin (North China
Pharmaceutical Group Corp., Shijiazhuang, China). The cells were
grown at 37.1°C in a 5% CO2 humidified atmosphere.
Analysis using a
3-(4,5-dimethyl-2-thiazolyl)-2,5-di- phenyl-2H-tetrazolium-bromide
(MTT) assay
The breast carcinoma cells were seeded at a density
of 7×103 cells/well in a 96-well plate for 24 h, and
were then treated with increasing doses of LCL161. Following 24, 48
and 72 h of exposure to LCL161, the cells were incubated with 15
µl MTT (5 mg/ml in PBS) per well for 4 h at 37.1°C. After 4
h, the supernatant was discarded and 150 µl of DMSO was
added to each well. The absorbance of the samples at 490 nm was
measured on a micro-plate reader (Synergy HT; BioTek Instruments,
Inc., Winooski, VT, USA).
Western blot analysis
The cells were collected and homogenized in
radioimmunoprecipitation assay lysis buffer (Roche Diagnostics,
Basel, Switzerland) for 0.5 h on ice. Subsequently, the cell
lysates were centrifuged at 13,225 × g for 0.5 h at 4°C to separate
the proteins. The proteins were quantified using a Bicinchoninic
Acid Protein Assay kit (BioSharp). The proteins (50 µg) were
dissociated via 10% SDS-PAGE and subsequently blotted onto
polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA,
USA) using Rotiphorese SDS-PAGE buffer (Carl Roth, Karlsruhe,
Germany). The membranes were blocked with skimmed milk (50
mg·l−1) for 2 h and incubated with the appropriate
primary antibody overnight at 4°C, followed by incubation with
conjugated secondary antibodies for 2 h at 20°C. The proteins were
visualized using enhanced chemiluminescence (ECL) reagents (EMD
Millipore) and the signals were measured using a Chemidoc XRS
imaging system (Universal Hood II; BioRad Laboratories, Inc.,
Hercules, CA, USA).
Colony formation assays
The cells (5×103 MDA-MB-231 cells;
7×103 MCF-7 cells) were seeded at the same density in
six-well plates for 24 h at 37°C. The cells were exposed to
different doses of LCL161. Cells were grown for 4 or 6 days at
37°C, and fixed with ECL reagents at −20°C for 10 min, following
which the colonies were stained with crystal violet (Beyotime
Institute of Biotechnology, Shanghai, China). Groups of cells
containing >50 cells were identified as a colony under a
microscope (CK30; Olympus Corporation, Toyko, Japan).
Mitochondrial transmembrane potential
(ΔΨm)
The detection of ΔΨm was performed using a
fluorescence microscope (IX71; Olympus Corporation) with
5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine
iodide (JC-1; Beyotime Institute of Biotechnology) staining.
Briefly, following drug treatment, the cells were incubated with 10
µmol·l−1 JC-1 for 30 min at 37°C in the dark.
Images of the cells were then captured using the IX71 Olympus
microscope.
Annexin V-FITC/PI apoptosis assay
Cellular apoptosis analysis was performed using an
Annexin V-FITC Apoptosis Detection kit (KeyGen Biotech Co., Ltd.).
The cells (2×105) were plated in a six-well plate and
treated with LCL161 for 24 h. The cells were harvested and
collected by low-speed centrifugation at 110.2 × g for 5 min at
25°C (LD5-2A; Beijing Lab Centrifuge Co., Ltd., Beijing, China) and
resuspended in 500 ml ice-cold binding buffer. Subsequently, the
cells were incubated with 5 µl Annexin V-FITC and PI for 15
min at room temperature in the dark, following which analysis was
performed using an Accuri C6 flow cytometer (BD Biosciences).
Electron microscopic detection
The cells from the various treatment groups were
fixed with 3% glutaraldehyde and 2% paraformaldehyde in 0.1 M PBS
buffer (pH 7.4) for 0.5 h, postfixed with 1% osmium tetroxide for
1.5 h, and washed and stained in 3% aqueous uranyl acetate for 1 h.
The samples were then dehydrated in an ascending series of ethanol
and acetone, and embedded in Araldite. Ultrathin sections were cut
on a Reichert ultramicrotome, double-stained with 0.3% lead
citrate, and examined under a JEM-1230 electron microscope (JEOL,
Ltd., Tokyo, Japan).
Small interfering (si)RNA
The breast cancer cells were seeded at a density of
2×105 cells/well in six-well plates and allowed to reach
~50% confluence on the day of transfection. A negative control RNA
construct (5′-UUC UCC GAA CGU GUC ACG UTT-3′) and siRNA against
RIP1 (5′-CCU UCU GAG CAG CUU GAU UTT-3′) were synthesized by
Shanghai GenePharma Co., Ltd. (Shanghai, China). The cells were
transfected with 20 nmol·l−1 siRNA in Opti-MEM medium
(Invitrogen; Thermo Fisher Scientific, Inc.) using Lipofectamine
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The
efficiency of siRNA knockdown was assessed using western blot
analysis 24 h following transfection.
Statistical analysis
The data are expressed as the mean ± standard error
of the mean from triplicate assays, and differences between
treatment groups were determined using two-tailed Student's
t-test. Statistical analysis was performed using Prism 5.0
software (GraphPad Prism, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
LCL161 inhibits the proliferation of
breast cancer cells
In order to determine the antitumor effect of LCL161
on breast cancer cells, the present study performed an MTT assay
24, 48 and 72 h following exposure of the cells to LCL161. As shown
in Fig. 1A, cell viability was
inversely proportional to the duration of exposure and the
concentration of LCL161 in the MDA-MB-231 and MCF-7 cells. When the
concentration of drug was increased (a to d), the number of cell
colonies was reduced. Colony outgrowth assays performed following
treatment with lower half maximal inhibitory concentrations in the
two cell lines also confirmed the inhibitory effects of LCL161 on
cell proliferation (Fig. 1B).
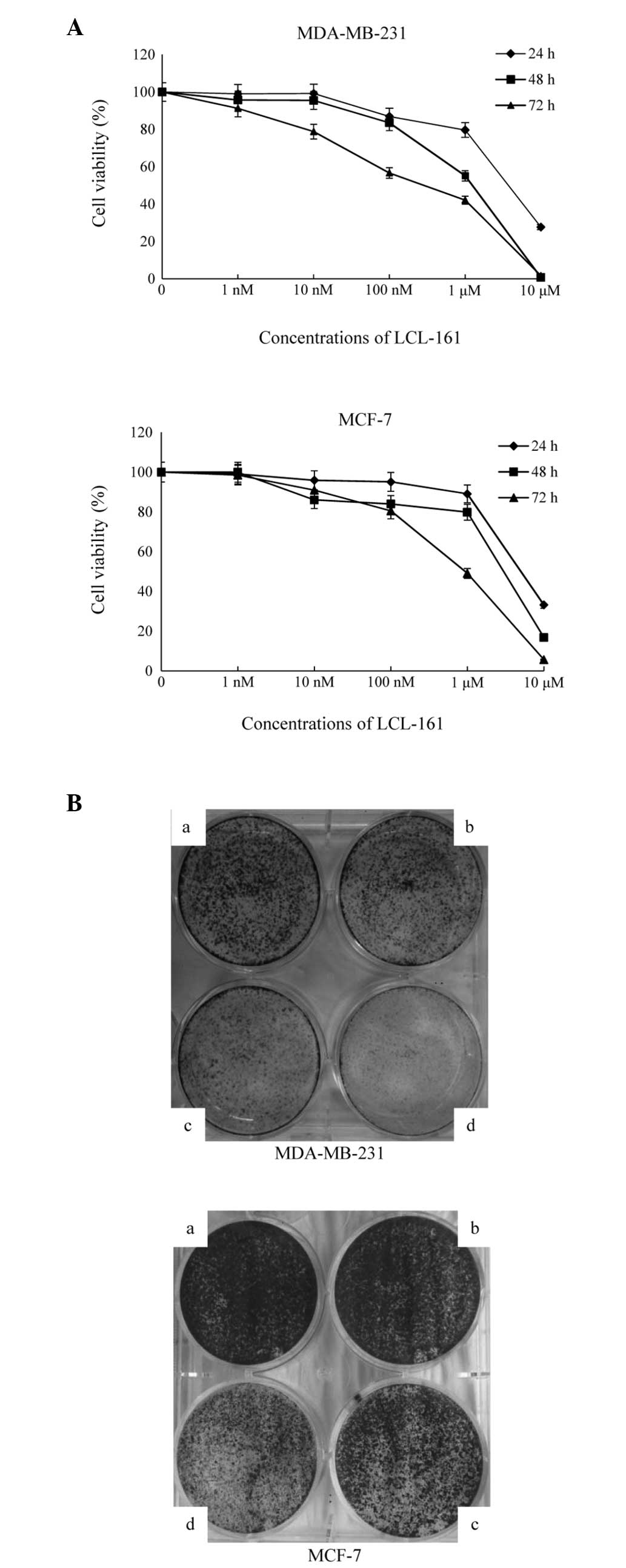 | Figure 1LCL161 inhibits the proliferation of
breast cancer cells. (A) MDA-MB-231 and MCF-7 cells were cultured
with various concentrations of LCL-161 (1, 10 and 100
nmol·l−1, 1 and 10 µmol·l−1) for 24,
48 and 72 h, following which cell viability was analyzed using a
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
assay. Data are presented as the mean ± standard error of the mean
from three independent experiments. (B) MDA-MB-231 cells were
treated with control, 2.5, 5 or 10 nmol·l−1 for 4 days,
and MCF-7 cells were treated with control, 10, 20 and 40
nmol·l−1 for 6 days. Colonies were stained with crystal
violet. Images are representative of three independent
experiments. |
LCL161 causes cell death via the
degradation of cIAP1 in breast cancer cells
To investigate whether modulation of IAPs occurred
following treatment with LCL161, the present study examined the
expression levels of the three major members of the IAPs family,
XIAP, cIAP1 and cIAP2, in the breast cancer cells. The results
revealed that the levels of cIAP1 were reduced in the two cell
lines, however, the effect was more marked in the MDA-MB-231.
LCL161 caused a marginal reduction in the levels of XIAP, and had
almost no effect on the levels of cIAP2 (Fig. 2). These results indicated that the
LCL161-dependent inhibition of proliferation was associated with
the degradation of cIAP1.
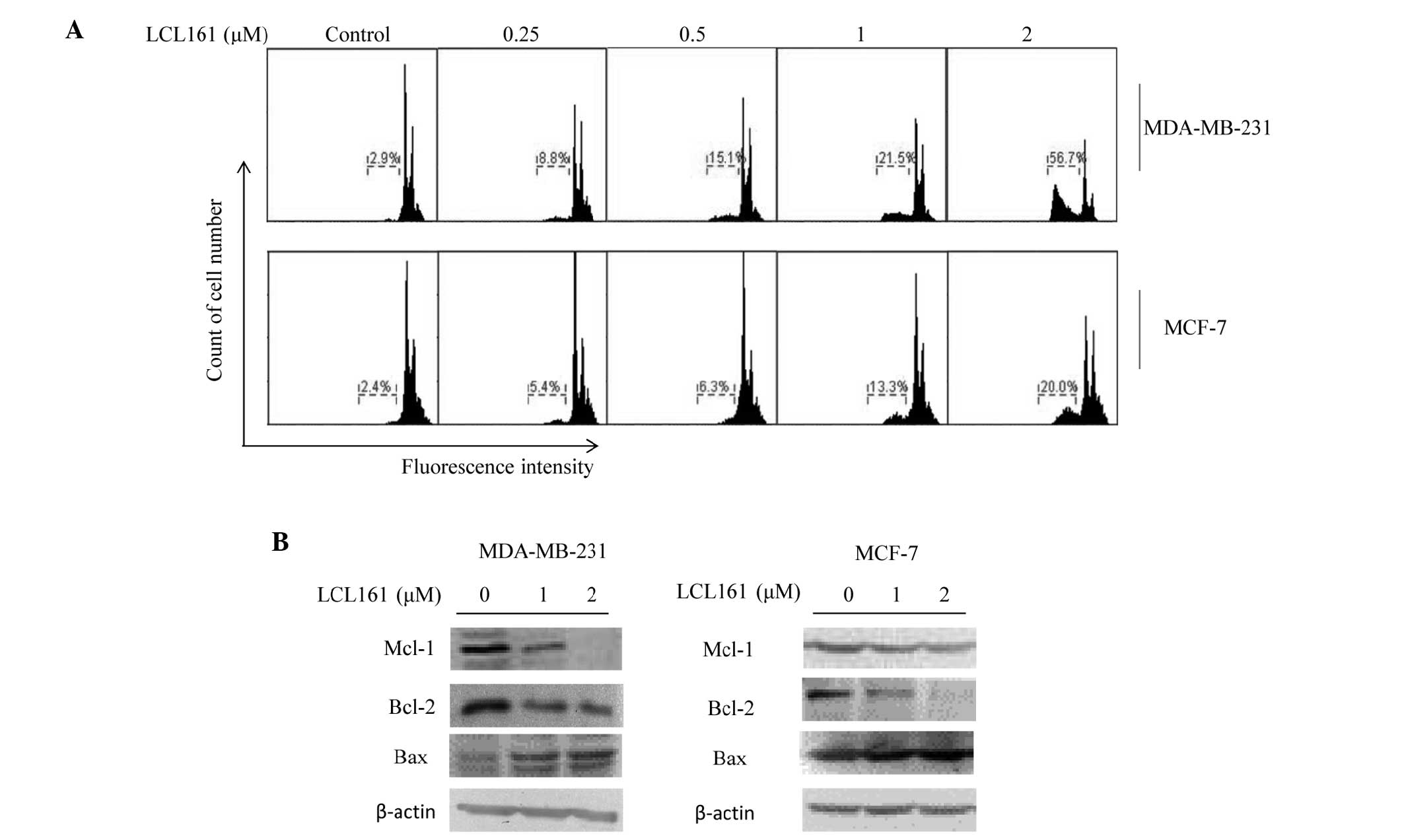
LCL161 induces apoptosis in breast cancer
cell lines
Flow cytometric analysis revealed that, in the
MDA-MB-231 cells, LCL161 induced apoptosis in a dose-dependent
manner, in which levels increased between 2.9 and 56.7%. Consistent
with the results of the MTT assay, the level of apoptosis in the
MCF-7 cells was lower, increasing between 2.4 and 20.0% (Fig. 3A). The present study also examined
the levels of several other apoptosis-associated proteins. Among
these, the expression levels of Mcl-1 and Bcl-2 were downregulated
following 24 h treatment with LCL161, whereas the expression of BAX
increased (Fig. 3B).
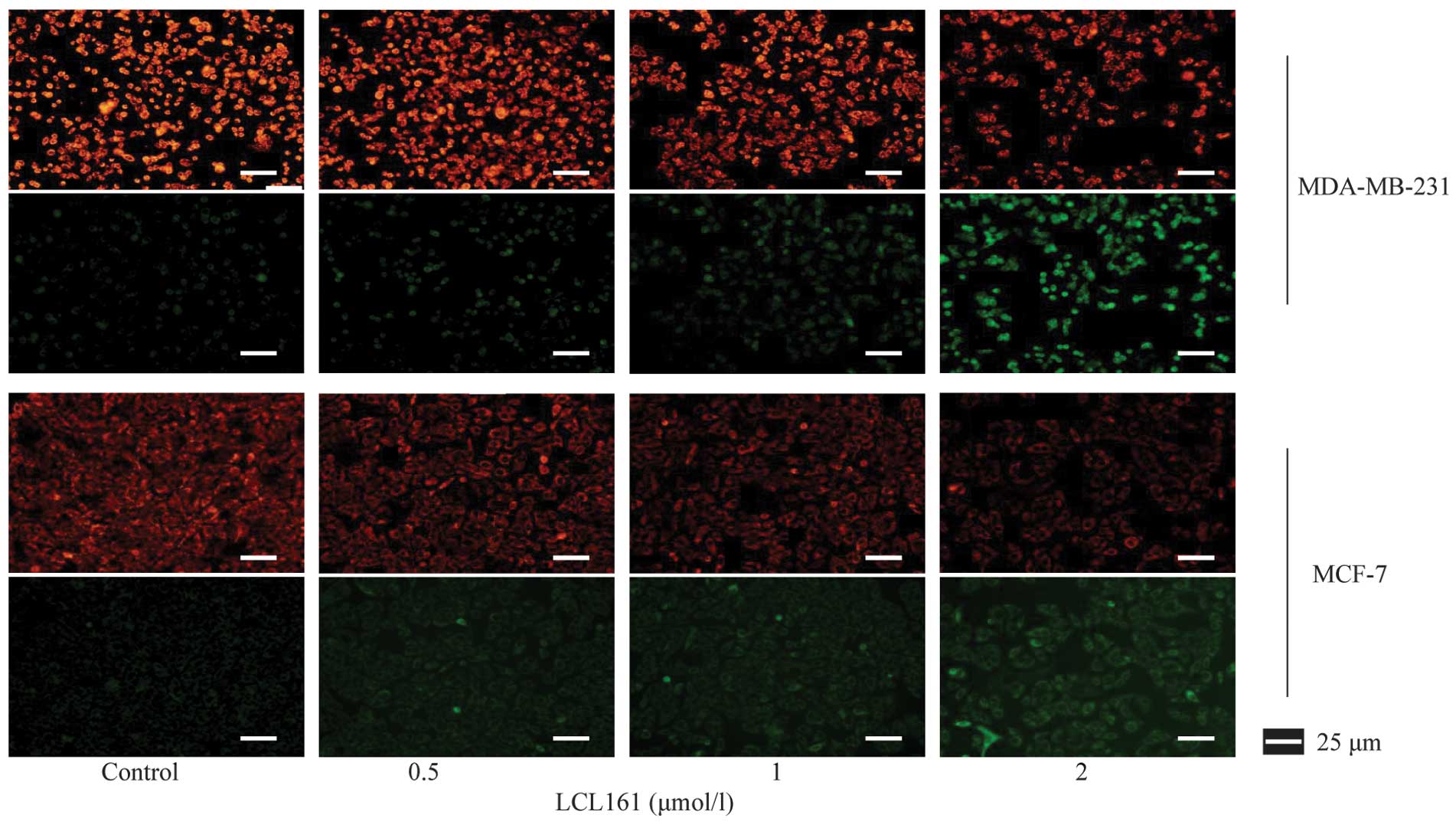
Fluorescence microscopy of the JC-1 stained cells
revealed that LCL161 reduced the ΔΨm, as there was a reduction in
the red/green ratio due to the loss of red fluorescent J aggregates
(Fig. 4). It was concluded from
these data that LCL161 effectively inhibited cell proliferation,
owing to its ability to activate the apoptotic pathway.
LCL161 in combination with pan-caspase
inhibitor induces necroptosis
To examine whether LCL161-induced apoptosis was
caspase-dependent, the MDA-MB-231 and MCF-7 cells were treated
separately with LCL161 in combination with Z-VAD-fmk, a cell
permeable pan-caspase inhibitor. As shown in Fig. 5A, Z-VAD-fmk treatment exacerbated
LCL161-dependent cell death. To further characterize this
caspase-independent cell death, cell morphology was examined using
transmission electron microscopy (TEM). As shown in Fig. 5B, in the breast cancer cells
treated with DMSO, Z-VAD or the Nec-1 RIP1 inhibitor, the majority
of cells exhibited a normal 'viable' morphology, which included
intact cytoplasmic membranes. By contrast, treatment with LCL161
alone induced cell death with classical apoptotic morphology. Under
the microscope, the chromatin was pyknotic and clotted, and
vacuolization due to the fusion of the endoplasmic reticulum and
plasma membrane appeared in the cytoplasm. The cytoplasmic
membranes were intact. By contrast, in the cells treated with the
Smac mimetic and Z-VAD in combination, the mitochondria were
swollen and cytoplasmic membranes were discontinuous; which are
hallmarks of necrosis. Thus, the form of cell death induced by
LCL161 in the absence of classical apoptosis appears to proceed via
a type of necrosis. In the cells treated with the combination of
LCL161 and the specific RIP1 inhibitor, Nec-1, the major form of
cell death was consistent with the cells treated with LCL161 alone
under TEM.
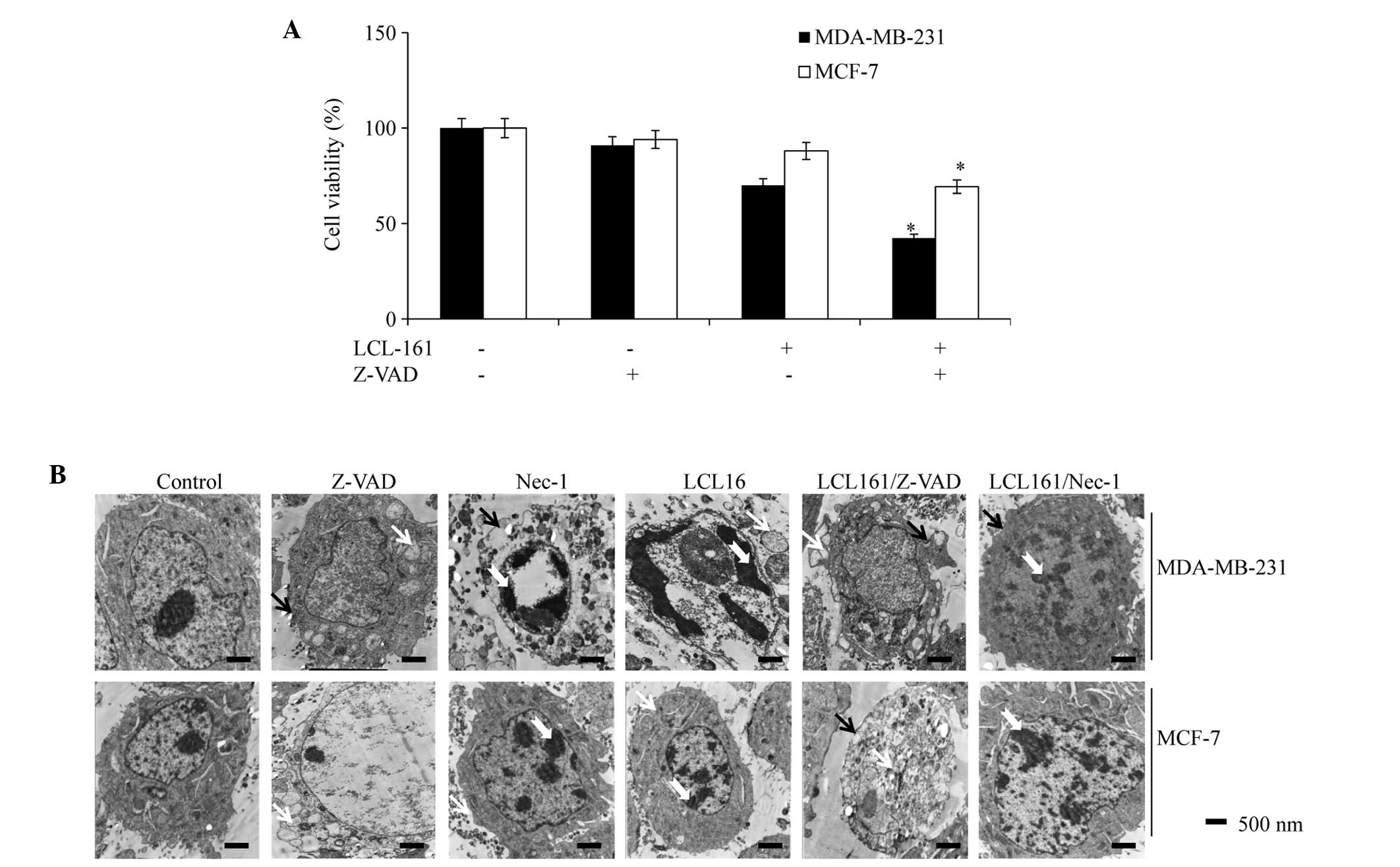 | Figure 5LCL-161+pan-caspase inhibitor
enhances cell death. (A) Viability of MDA-MB-231 or MCF-7 cells
treated with DMSO, LCL161 (1 µmol·l−1) and
LCL161/z-VAD (20 µmol·l−1) were analyzed using a
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
assay. Data are representative of three independent experiments and
are expressed as the mean ± standard error of the mean
(*P<0.05, vs. the LCL161 group). (B) MDA-MB-231 and
MCF-7 cells were treated with DMSO, z-VAD, Nec-1, LCL161,
LCL161+z-VAD or LCL161+Nec-1 and were analyzed by electron
microscopy. White arrowheads indicate the swelling of cellular
organelles in the cells treated with z-VAD or LCL161+z-VAD; black
arrowheads indicate cell membrane integrity in the cells treated
with DMSO, Nec-1, LCL161 or LCL161+Nec-1, and membrane breakdown in
the cells treated with z-VAD or LCL161+z-VAD. Dovetail arrowheads
indicate pyknosis and clotted chromatin. DMSO, dimethyl sulfoxide;
Nec-1; necrostatin-1. |
Cell death induced by the combination of
LCL161 and Z-VAD-fmk is dependent on RIP1
An Annexin V-FITC/PI apoptosis assay was used to
detect the levels of apoptosis induced by LCL161 with Z-VAD and/or
Nec-1 on the MDA-MB-231 and MCF-7 cells. The results revealed that
the apoptotic rate decreased in the LCL161/Z-VAD group, however,
Nec-1 inhibited this increase, resulting in a lower apoptotic rate,
compared with the LCL161-only group (Fig. 6A). In addition, the results
demonstrated that the viability of the MDA-MB-231 cells following
treatment with Nec-1 and LCL161 was similar to that following
LCL161 treatment alone (81 and 78%, respectively; Fig. 6B) These results suggested that
LCL161+Z-VAD-induced necroptosis was regulated by Nec-1.
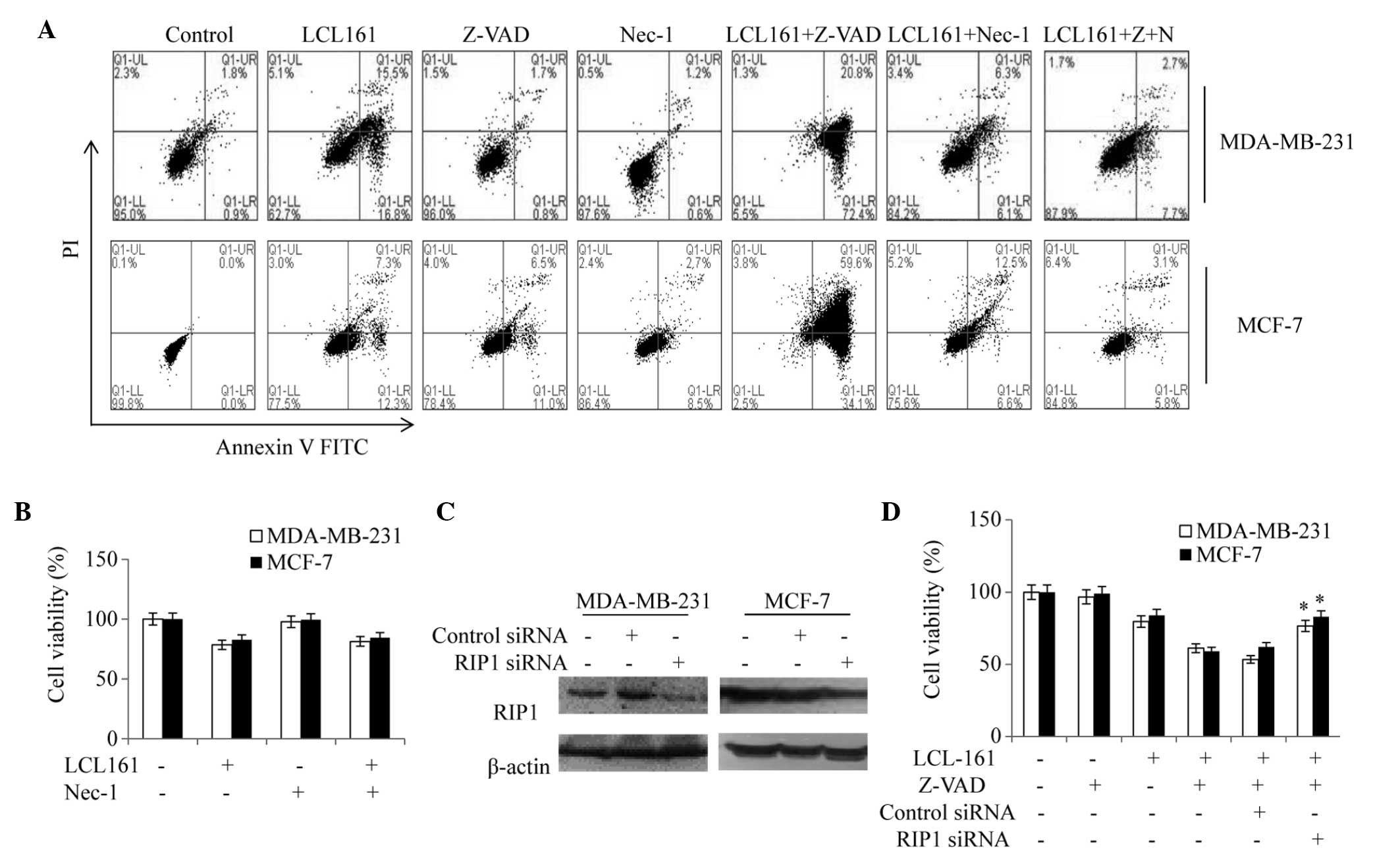 | Figure 6Cell death induced by
LCL161+z-VAD-fmk depends on RIP1. (A) MDA-MB-231 and MCF-7 cells
treated with DMSO, z-VAD, Nec-1, LCL161, LCL161+z-VAD, and
LCL161+Nec-1 for 24 h were analyzed using an Annexin V-FITC/PI
apoptosis assay. (B) MDA-MB-231 and MCF-7 cells treated with DMSO,
LCL161, Nec-1 and LCL161+Nec-1 for 24 h were analyzed using a
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
assay. (C) MDA-MB-231 and MCF-7 cells were transfected with control
or RIP1 siRNA and, after 48 h, whole-cell lysates were subjected to
western blot analysis. (D) MDA-MB-231 and MCF7 cells were
transfected with RIP1 siRNA, cultured for 24 h and then treated
with LCL161 or LCL161+z-VAD. Data are representative of three
independent experiments and are expressed as the mean ± standard
error of the mean (*P<0.05, vs. the LCL161+Z-VAD
group). RIP1, receptor-interacting protein kinas-1; DMSO, dimethyl
sulfoxide; Nec-1; necrostatin-1; FITC, fluorescein isothiocyanate;
PI, propidium iodide; siRNA, small interfering RNA; UR, upper right
(late apoptotic cells); UL, upper left (cell debris); LL, lower
left (normal cells); LR, lower right (early apoptotic cells). |
The present study also examined whether specific
ablation of the expression of RIP1 by siRNA phenocopied the effect
of the RIP1 inhibitor. Notably, the downregulation of RIP1
protected the cells, compared with the cells treated with LCL161
alone (Fig. 6C and D), confirming
that necroptosis induced by the combination of LCL161 and Z-VAD-fmk
was dependent on RIP1.
Discussion
Clinically, breast cancer can be divided into three
major subclasses: Luminal A/B subtype, HER-2-amplified subtype, and
basal-like subtype (7). Treatment
with trastuzumab, an anti-HER2 monoclonal antibody, improves the
overall survival rate of patients with HER2-positive breast cancer
in adjuvant and first-line settings (29). However, no therapy has demonstrated
such efficacy in the treatment of the other two breast cancer
subtypes. Thus, the present study selected two breast carcinoma
lines of the non HER-2-amplified subtype in order to evaluate the
efficacy of the Smac mimetic, LCL161.
The results of the present demonstrated that the
Smac mimetic, LCL161, induced apoptosis and inhibited the
proliferation of the breast cancer cell lines. Apoptosis is a
strictly regulated process, and its misregulation is involved in
several human diseases, including cancer (30). As key regulators of apoptosis, IAPs
bind to and inhibit key caspases, thus conferring resistance to
several treatment regimens (31,32).
LCL161 is a newly designed orally bioavailable monovalent mimetic
of Smac, which has advanced in clinical development. It was
designed to mimic the AVPI tetrapeptide binding motif of Smac,
therefore, it can interact with XIAP, cIAP1 and cIAP2 (33). Oral administration of LCL161
inhibits tumor growth in a mouse model of multiple myeloma
(34). In breast carcinoma cells,
the reduction of cIAP1 following exposure to LCL161 may be due to
the binding of LCL161 to cIAP1. The binding leads to a
conformational change of cIAP1, stimulating RING domain-dependent
homodimerization, which results in cIAP1 auto-ubiquitination and
subsequent rapid proteasomal degradation. Upon loss of cIAP1, the
non-ubiquitinated form of RIP1, together with FADD and caspase-8
form a complex that activates caspase-8 and triggers the extrinsic
pathway of apoptosis (34).
Bcl-2 family protein members are indispensable for
the correct function of major organ systems, and mutations
affecting their levels or activity are correlated with cancer
(35). These proteins are either
pro-apoptotic, including BAX and Bcl-2 antagonist/killer 1 (BAK),
or anti-apoptotic, including Bcl-2, Mcl-1, Bcl-extra large (XL) and
Bcl-W). The activity of these proteins ultimately converges on the
mitochondrial outer membrane, and an excess of pro-apoptotic
activity triggers mitochondrial membrane permeabilization and
caspase activation (36). There is
extensive crosstalk between the mitochondrial and death
receptor-mediated death pathway. For example,
BH3-interacting-domain death agonist (BID), a pro-apoptotic Bcl-2
member, which is processed to truncated BID (tBID) by the activity
of caspase-8. tBID translocates to the mitochondrial membrane,
where it binds to BAX and BAK, and stimulates the release of
cytochrome c, leading to activation of the intrinsic pathway
(37). By contrast, anti-apoptotic
MCL-1 is integral in cell survival and apoptosis (38,39).
The present study assessed the effects of LCL161 on Mcl-1, BAX and
BAK. The compound reduced the level of Mcl-1, and concomitantly
increased the levels of BAX and BAK, aggravating the mitochondrial
pathway of apoptosis.
However, the present study also observed a
caspase-independent, RIP1-dependent, type of necrotic cell death in
response to LCL161. This indicates another example of necroptosis,
which occurs when caspases are inhibited or when apoptosis cannot
be activated efficiently. Since cIAP1 is an E3 ubiquitin ligase
(40,41), its loss upon LCL161 treatment leads
to the accumulation, or reduced ubiquitination, of cIAP substrates,
which may include RIP1, a known target of XIAP and cIAPs (42). Thus, the present study hypothesized
that LCL161-dependent degradation of cIAP1 leads to the
accumulation of active RIP1, which in turn triggers formation of
the necrosome and cell death.
In conclusion, the findings of the present study
suggested that LCL161 may be suitable for application as a targeted
therapeutic for the treatment of patients with breast cancer.
Future investigations are requireded to determine the efficacy and
therapeutic index of LCL161 in vivo.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81372899). The authors would
like to thank Editage/Cactus for assistance with English language
editing.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Bray F, Ren JS, Masuyer E and Ferlay J:
Global estimates of cancer prevalence for 27 sites in the adult
population in 2008. Int J Cancer. 132:1133–1145. 2013. View Article : Google Scholar
|
|
3
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Higgins MJ and Baselga J: Targeted
therapies for breast cancer. J Clin Invest. 121:3797–3803. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bourdeanu L and Luu T: Targeted therapies
in breast cancer: Implications for advanced oncology practice. J
Adv Pract Oncol. 5:246–260. 2014.
|
|
9
|
Reed JC: Apoptosis-based therapies. Nat
Rev Drug Discov. 1:111–121. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: A link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lockshin RA and Williams CM: Programmed
cell Death-I. Cytology of degeneration in the intersegmental
muscles of the pernyi silkmoth. J Insect Physiol. 11:123–133. 1965.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bai L, Smith DC and Wang S: Small-molecule
SMAC mimetics as new cancer therapeutics. Pharmacol Ther.
144:82–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dubrez L, Berthelet J and Glorian V: IAP
proteins as targets for drug development in oncology. Onco Targets
Ther. 9:1285–1304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Du C, Fang M, Li Y, Li L and Wang X: Smac,
a mitochondrial protein that promotes cytochrome c-dependent
caspase activation by eliminating IAP inhibition. Cell. 102:33–42.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Salvesen GS and Duckett CS: IAP proteins:
Blocking the road to death's door. Nat Rev Mol Cell Biol.
3:401–410. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, McQuade T, Siemer AB, Napetschnig J,
Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et
al: The RIP1/RIP3 necrosome forms a functional amyloid signaling
complex required for programmed necrosis. Cell. 150:339–350. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Holler N, Zaru R, Micheau O, Thome M,
Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp
J: Fas triggers an alternative, caspase-8-independent cell death
pathway using the kinase RIP as effector molecule. Nat Immunol.
1:489–495. 2000. View
Article : Google Scholar
|
|
19
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smith CC, Davidson SM, Lim SY, Simpkin JC,
Hothersall JS and Yellon DM: Necrostatin: A potentially novel
cardioprotective agent? Cardiovasc Drugs Ther. 21:227–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Linkermann A, Bräsen JH, Himmerkus N, Liu
S, Huber TB, Kunzendorf U and Krautwald S: Rip1
(receptor-interacting protein kinase 1) mediates necroptosis and
contributes to renal ischemia/reperfusion injury. Kidney Int.
81:751–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia-reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin J, Li H, Yang M, Ren J, Huang Z, Han
F, Huang J, Ma J, Zhang D, Zhang Z, et al: A role of RIP3-mediated
macrophage necrosis in atherosclerosis development. Cell Rep.
3:200–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen KF, Lin JP, Shiau CW, Tai WT, Liu CY,
Yu HC, Chen PJ and Cheng AL: Inhibition of Bcl-2 improves effect of
LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem
Pharmacol. 84:268–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Varfolomeev E, Blankenship JW, Wayson SM,
Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO,
Wallweber HJ, et al: IAP antagonists induce autoubiquitination of
c-IAPs, NF-kappaB activation and TNFalpha-dependent apoptosis.
Cell. 131:669–681. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weisberg E, Ray A, Barrett R, Nelson E,
Christie AL, Porter D, Straub C, Zawel L, Daley JF, Lazo-Kallanian
S, et al: Smac mimetics: Implications for enhancement of targeted
therapies in Leukemia. Leukemia. 24:2100–2109. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marty M, Cognetti F, Maraninchi D, Snyder
R, Mauriac L, Tubiana-Hulin M, Chan S, Grimes D, Antón A, Lluch A,
et al: Randomized phase II trial of the efficacy and safety of
trastuzumab combined with docetaxel in patients with human
epidermal growth factor receptor 2-positive metastatic breast
cancer administered as first-line treatment: The M77001 study
group. J Clin Oncol. 23:4265–4274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lowe SW and Lin AW: Apoptosis in cancer.
Carcinogenesis. 21:485–495. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang S, Bai L, Lu J, Liu L, Yang CY and
Sun H: Targeting inhibitors of apoptosis proteins (IAP) for new
breast cancer therapeutics. J Mammary Gland Biol Neoplasia.
17:217–228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. Biomed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu G, Chai J, Suber TL, Wu JW, Du C, Wang
X and Shi Y: Structural basis of IAP recognition by Smac/DIABLO.
Nature. 408:1008–1012. 2000. View
Article : Google Scholar
|
|
34
|
Chauhan D, Neri P, Velankar M, Podar K,
Hideshima T, Fulciniti M, Tassone P, Raje N, Mitsiades C, Mitsiades
N, et al: Targeting mitochondrial factor Smac/DIABLO as therapy for
multiple myeloma (MM). Blood. 109:1220–1227. 2007. View Article : Google Scholar
|
|
35
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kvanskul M and Hinds MG: Structural
biology of the Bcl-2 family and its mimicry by viral proteins. Cell
Death Dis. 4:e9092013. View Article : Google Scholar
|
|
37
|
Kim BM and Chung HW: Desferrioxamine (DFX)
induces apoptosis through the p38-caspase8-Bid-Bax pathway in
PHA-stimulated human lymphocytes. Toxicol Appl Pharmacol.
228:24–31. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kozopas KM, Yang T, Buchan HL, Zhou P and
Craig RW: MCL1, a gene expressed in programmed myeloid cell
differentiation, has sequence similarity to BCL2. Proc Natl Acad
Sci USA. 90:3516–3520. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sano M, Hayashi E, Murakami H, Kishimoto
H, Fukuzawa R and Nemoto N: Mcl-1, an anti-apoptotic Bcl-2 family
member, essentially overlaps with insulin-producing cells in
neonatal nesidioblastosis. Virchows Arch. 452:469–470. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gyrd-Hansen M, Darding M, Miasari M,
Santoro MM, Zender L, Xue W, Tenev T, da Fonseca PC, Zvelebil M,
Bujnicki JM, et al: IAPs contain an evolutionarily conserved
ubiquitin-binding domain that regulates NF-kappaB as well as cell
survival and oncogenesis. Nat Cell Biol. 10:1309–1317. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vaux DL and Silke J: IAPs, RINGs and
ubiquitylation. Nat Rev Mol Cell Biol. 6:287–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park SM, Yoon JB and Lee TH: Receptor
interacting protein is ubiquitinated by cellular inhibitor of
apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett.
566:151–156. 2004. View Article : Google Scholar : PubMed/NCBI
|