Introduction
Breast cancer is the most common type of cancer
diagnosed in American women and is the second most common cause of
cancer-associated mortality. In 2014, 232,670 new breast cancer
cases are expected to be diagnosed, and 40,430 breast cancer
mortalities are expected in the USA (1). In the treatment of breast cancer,
anthracyclines (e.g. doxorubicin and daunorubicin) are among the
most active anti-neoplastic agents in use (2–5).
Although highly effective in chemotherapy, the mechanism of action
of these compounds is not specific and several mechanisms have been
demonstrated, including inhibition of topoisomerase I/II and
production of reactive oxygen species (ROS). In addition, clinical
use of anthracyclines is often hampered by its cumulative
dose-limiting cardiotoxicity, as well as the development of drug
resistance by tumor cells (6,7).
Following the success of doxorubicin, a tremendous amount of effort
has been made in the search of effective alternatives; however,
only few of them are clinically approved and none of them are
clearly superior to doxorubicin (8,9).
Therefore, the development of effective therapeutic strategies,
including alternatives to anthracyclines and/or their combination
with other effective agents, remains an important task in breast
cancer treatment.
The anti-cancer agent amsacrine (m-AMSA; Fig. 1) is a topoisomerase II inhibitor
and is structurally a 9-anilinoacridine analogue.
9-Anilinoacridines have displayed an advantage over other
topoisomerase II inhibitors in that they were not affected by
transport-mediated multidrug resistance (MDR). In addition,
atypical MDR can be overcome by structure modification at its
topoisomerase II-binding domain (10). In an attempt to develop improved
chemotherapeutic agents, 9-anilinoacridine has been subjected to
various structural modifications on the acridine backbone as well
as on the aniline ring. It is generally thought that the mode of
action of this type of compound is through DNA intercalations
facilitated by the tricyclic acridine moiety; additional
interactions between the aniline ring of the compound with other
molecules can lead to the inhibition of topoisomerase II (11,12).
A previous study by our group reported the synthesis
and preliminary anti-cancer evaluation of a series of
sulfur-containing 9-anilinoacridine derivatives, including
2-({4-[4-(acridin-9-ylamino) phenylthio]phenyl}
(2-hydroxyethyl)amino)ethan-1-ol (CK0402) and
3-({4-[4-(acridin-9-ylamino)
phenylthio]phenyl}(3-hydroxypropyl)amino)propan-1-ol (CK0403)
(Fig. 1) (13). Within a library of
sulfur-containing 9-anilinoacridines, CK0403 and CK0402 were the
most cytotoxic agents against Chinese hamster lung transformed V79
cells (13). Similarly to
amsacrine and anthracyclines, CK0403 inhibited a topoisomerase
IIα-catalyzed decatenation reaction, and it was ~10 times more
effective than amsacrine and its analogue CK0402 according to study
by our group (14). A further
study by our group showed that CK0402 was effective against
numerous breast cancer cell lines; in addition, the combination of
CK0402 with herceptin enhanced its activity in HER2(+) SKBR-3 cells
(15). Reduced production of ROS
was noted in CK0403-treated fibroblast cells compared with that in
cells treated with CK0402; furthermore, the cell proliferation
inhibitory activity of CK0403 was enhanced when combined with
NU1025, a potent and specific poly(adenosine diphosphate-ribose
polymerase 1 inhibitor (14). In
addition, a clonogenic assay demonstrated that CK0403 was more
potent than CK0402 against hepatocellular carcinoma HepG2 cells;
however, in breast cancer MCF-7 cells, the activity of CK0403 and
CK0402 was comparable (14).
To examine whether CK0403 is more active than CK0402
against breast cancer cell lines other than MCF7, the present study
first evaluated the cytotoxic effect of CK0403 on a panel of
established human breast cell lines with varying levels of estrogen
receptor (ER), progesterone receptor (PR) and HER2/neu, the
most common clinically used biomarkers for breast cancer treatment.
The human breast cell lines used included MCF-7 [ER(+), HER2(−) and
PR(+)], BT-474 [ER(+), HER2 overexpressing and PR(+)], MDA-MB-231
[ER(−), HER2(−), PR(−)] and SKBR-3 [ER(−), HER2 overexpressing,
PR(−)], and the non-cancerous MCF-10A cell line [ER(−), HER2(−),
PR(−)]. The cytotoxicity of CK0403 was subsequently evaluated on
the most sensitive cell line, SKBR-3, under hypoxic conditions or
with the combination of herceptin. The potential underlying
mechanisms of the cytotoxic effects of CK0403 were also explored
using cell cycle analysis, quantification of the apoptotic rate and
assessment of autophagy-associated signaling.
Materials and methods
Chemicals and reagents
CK0403 was prepared and purified as described
previously (13). Herceptin was
kindly provided by Genentech Inc. (San Francisco, CA, USA).
Dimethyl sulfoxide (DMSO, cell culture grade), sulforhodamine B and
propidium iodide were purchased from Sigma-Aldrich Chemical Co.
(St. Louis, MO, USA). Dulbecco's modified Eagle's medium
(DMEM)/F12, Iscove's modified Dulbecco's medium (IMDM), horse
serum, penicillin/streptomycin, phosphate-buffered saline (PBS),
RNase A and trypsin-EDTA were purchased from Gibco-BRL (Invitrogen
Life Technologies, Inc. Carlsbad, CA, USA). Fetal bovine serum
(FBS) was purchased from Gemini Bio-Products (Woodland, CA,
USA).
Cell culture and treatment
All of the cell lines were obtained from the
American Type Culture Collection (Manassas, VA, USA) and grown in
75-cm2 cell culture flasks at 37°C in a humidified
atmosphere of 5% CO2 in air. MCF-7 and MDA-MB-231 cells
were maintained in IMDM/F12 (1:1 mixture) medium; SKBR-3 and BT-474
cells were maintained in DMEM/F12 (1:1 mixture). All the media were
supplemented with penicillin/streptomycin (50 mg/ml) and 10% FBS.
MCF-10A cells were maintained in DMEM/F-12 supplemented with 5%
horse serum, insulin (10 mg/ml), hydrocortisone (500 ng/ml),
epidermal growth factor (20 ng/ml), cholera toxin (100 ng/ml) and
penicillin/streptomycin (50 mg/ml). For hypoxia treatment, cells
were incubated in 1% O2/5% CO2/94%
N2 overnight prior to drug treatment. Hypoxic conditions
were generated using a PROOX sensor (model C21; BioSpherix,
Hanover, NH, USA) using N2 influx. Drug treatment
involved continuous incubation with the compounds for six days. For
all cell culture experiments, cells were seeded at least 24 h prior
to treatment.
Cell proliferation assay
Cells (~3,000–5,000/well) were seeded and allowed to
grow in a 96-well plate for at least 24 h prior to treatment. DMSO
was used as a vehicle to dissolve CK0403 with a final DMSO
concentration ≤0.1% (v/v) in the medium. Herceptin was added 3 h
prior to the addition of CK0403 when cells were treated with the
combination of CK0403 and herceptin. At the end of the incubation,
cultures were fixed with 50 µl 50% cold trichloroacetic acid
and incubated at 4°C for 1 h. The plates were washed five times
with water and then air-dried. The fixed cells were stained for 30
min with 100 µl 0.4% sulforhodamine B solution in 1% acetic
acid. The plates were then washed five times with 1% acetic acid to
remove any unbound dye. The bound dye was dissolved with 10 mM Tris
buffer and the absorbance of the resulting solution was measured at
570 nm to quantify the number of surviving cells. All of the
treatments were performed at least three times in triplicate. The
drug concentration that produced a 50% reduction (LC50)
in cell survival was determined using median-effect plots.
Apoptotic cell death detection
To quantitatively determine the apoptotic cell death
induced by CK0403, CK0402 and amsacrine (Sigma-Aldrich Chemical
Co.), a cell death detection ELISA kit (Roche Diagnostics,
Indianapolis, IN, USA) was used and enrichment factor was reported,
according to the manufacturer's instructions. Briefly, following
treatment of cells with the indicated drugs at 0.3125 or 0.525
µM for 72 h, the cells were lysed and incubated for 30 min
at room temperature; aliquots of 20 µl supernatant were then
transferred into the wells of a streptavidin-coated microtiter
plate. To each well, 80 µl immunoreagent was added. After
incubation at room temperature for 2 h, the solution was decanted,
and each well was rinsed three times with incubation buffer. Color
development was performed by adding 100 µl
2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS)
solution and absorbency was measured at 405 nm in a microtiter
plate reader (Spectramax Plus 384; Molecular Devices, Sunnyvale,
CA, USA) against ABTS solution as a blank.
Flow cytometric cell cycle analysis
SKBR-3 cells growing in the exponential phase were
treated with the indicated concentrations of CK0403 or DMSO as
previously described (15). At the
end of the incubation, cells were harvested, fixed in ice-cold 70%
ethanol and stored at −20°C. The fixed cells were washed with
phosphate-buffered saline, treated with RNase A (3 units/ml) at
37°C for 30 min and stained with propidium iodide (50 mg/ml) for 5
min. DNA content for 250,000 cells per analysis was monitored using
a Becton-Dickinson FACScan flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA) and Modfit software (LT version 2.0; Verity
Software House, Topsham, ME) was used for analysis of the cell
cycle distribution.
Western blot analysis
SKBR-3 cells were treated with CK0403, CK0402,
amsacrine, doxorubicin or camptothecin (Sigma-Aldrich Chemical Co.)
at 1 µM for 24 h. Cells were harvested by scraping and
washed with PBS. Cellular proteins were isolated with cell lysis
buffer purchased from Cell Signaling Technology, Inc (Beverly, MA,
USA). Equal amounts of protein (40 µg) were boiled for 5
min, separated by 10% SDS-PAGE at 100 V for 110 min and then
electro-transferred onto polyvinylidene difluoride membranes.
Antibody against Atg5 was purchased from Novus (Littleton, CO, USA;
rabbit polyclonal; cat. no. NB110-53818; 1:3,000). β-Actin antibody
(as a reference standard) was purchased from Sigma-Aldrich (mouse
monoclonal; cat. no. A5316; 1:5,000). Primary antibody incubations
were performed at 4°C overnight. Following extensive washing,
specific bands were detected using immobilon western
chemiluminescent substrate (Millipore, Billerica, MA, USA).
Secondary anti-rabbit (cat. no. 7076) or anti-mouse (cat. no. 7074)
immunoglobulin G conjugated to horse radish peroxidase were
purchased from Cell Signaling Technology, Inc.
Statistical analysis
The statistical comparisons between certain pairs of
measurements were performed using Student's t-test. Statistical
analyses were performed using Microsoft Excel 2007. The data are
expressed as the mean ± standard deviation.
Results
CK0403 inhibits the growth of human
breast cancer cells
The human breast cancer cell lines MCF-7,
MDA-MB-231, BT-474 and SKBR-3 were selected to evaluate the growth
inhibitory activity of CK0403. Based on a previous study by our
group on CK0402, the cells were incubated with CK0403 for six days
(15). In addition, the growth
inhibitory effect of CK0403 on the non-cancerous human breast cell
line MCF-10A was examined. As shown in Table I, MCF-10A was the cell line with
the lowest sensitivity to CK0403 treatment. Among the breast cancer
cell lines tested, the magnitude of the growth inhibitory effects
of CK0403 was in the following order:
SKBR3≥MDA-MB-231>BT-474≥MCF-7.
 | Table ILC50 values of CK0403 in a
panel of breast cancer cell lines and the normal breast cell line
MCF10A. |
Table I
LC50 values of CK0403 in a
panel of breast cancer cell lines and the normal breast cell line
MCF10A.
| Cell line | LC
(µM)a 50 | Immunoprofile |
|---|
| MCF-7 | 1.06±0.35 | ER+,
HER2−, PR+ |
| MDA-MB-231 | 0.09±0.02 | ER−,
HER2−, PR− |
| BT-474 | 0.94±0.53 | ER+,
HER2+, PR+ |
| SKBR-3 | | ER−,
HER2+, PR− |
| Normoxia | 0.07±0.05 | |
| Hypoxia | 0.12±0.03 | |
| MCF10A | 4.25±0.93 | ER−,
HER2−, PR |
A previous study by our group reported that CK0402
induced apoptosis and autophagy in SKBR-3 ells (15). Autophagy is induced by a variety of
metabolic stresses, including hypoxia and oxidative stress. To
elucidate the role of autophagy in the cell death induced by
CK0403, the growth inhibitory activity of CK0403 in SKBR-3 cells
was examined under normoxic and hypoxic conditions in parallel. The
results showed that CK0403 was less potent under hypoxic conditions
as compared with normoxia (LC50=0.12 µM vs. 0.07
µM, respectively).
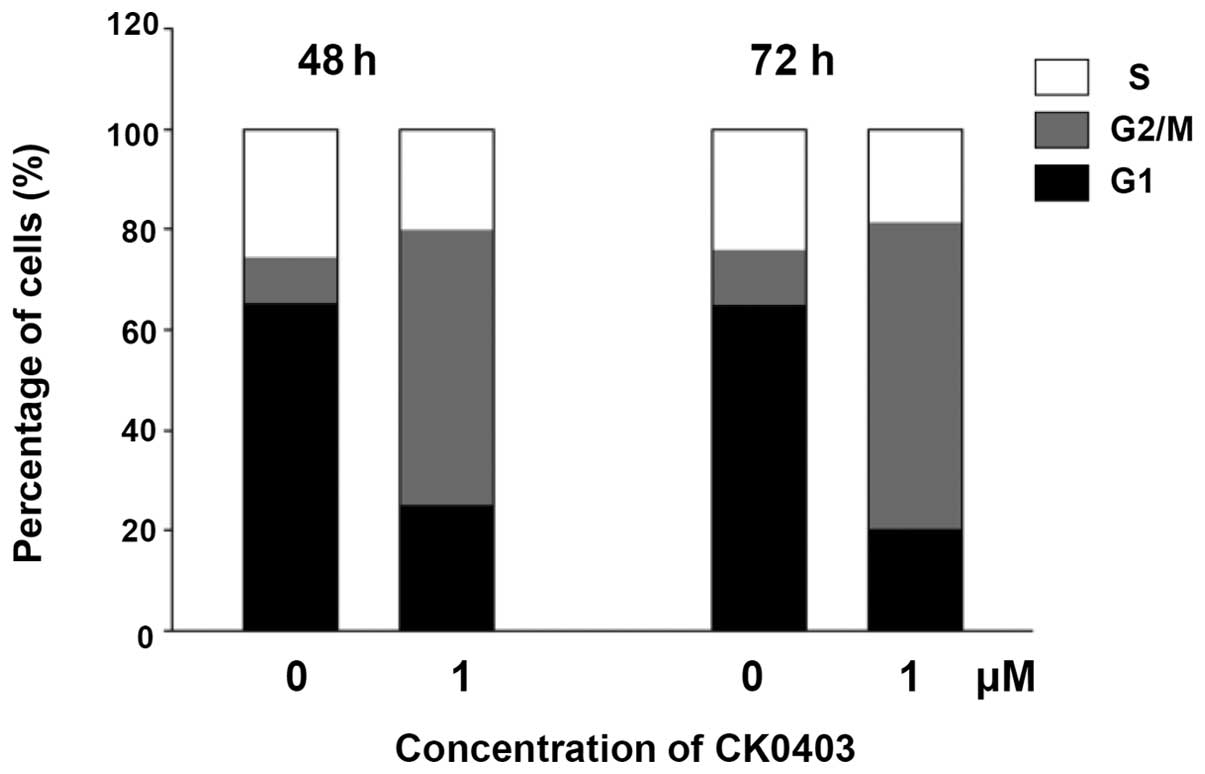
CK0403 induces G2/M phase
arrest in SKBR-3 cells
In order to examine the cell cycle distribution of
SKBR-3 cells, cells were treated with CK0403 at 1 µM for 48
or 72 h, stained with propidium iodide and subjected to flow
cytometric analysis (Fig. 2). The
results showed that CK0403 time-dependently induced G2/M
arrest with a reduced G1- and S-phase population in
SKBR-3 cells. These results are similar to those observed for
CK0402; however, it was noted that CK0403 induced a lower increase
in the G2/M-phase population as compared to CK0402.
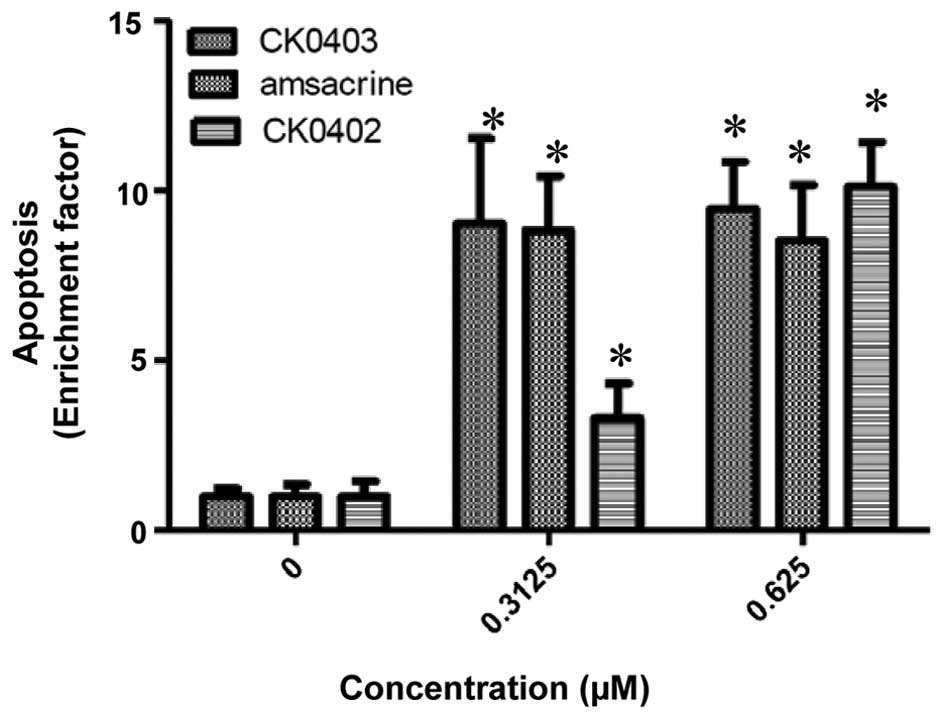
CK0403 induces apoptosis in SKBR-3 breast
cancer cells
The cell growth inhibition resulting from CK0403
treatment was further characterized by evaluating the apoptotic
response induced by CK0403 and compared with that of CK0402 and
amsacrine in SKBR-3 cells using an ELISA kit (Fig. 3) (15). As it was previously found that
CK0402 did not induce apoptosis effectively within 48 h of
treatment, apoptosis was analyzed at 72 h. The results showed that
CK0403 induced apoptotic effects in SKBR-3 cells at doses of 0.3125
and 0.625 µM, resulting in a 9.02±2.51- and 9.47±1.39-fold
enhancement of apoptosis, respectively. The results of the present
study showed that at the dose of 0.3125 µM, CK0403 induced
apoptosis in SKBR-3 cells more effectively than its analogue
CK0402, but equally effective as amsacrine. At the dose of 0.625
µM, all three compounds were equally effective at inducing
apoptosis in SKBR-3 cells.
CK0403 induces the autophagic pathway in
SKBR-3 cells
Atg5 cleavage provokes apoptotic cell death, and it
represents a molecular link between autophagy and apoptosis
(16). As shown in Fig. 4, the present study demonstrated
that treating SKBR3 cells with CK0403 at the dose of 1 µM
led to the induction of cleaved Atg5 protein expression.
Furthermore, it was shown that at the same dose, CK0403 induced
higher levels of cleaved Atg5 protein than other anti-cancer agents
tested, including CK0402, amsacrine, doxorubicin and
camptothecin.
Herceptin potentiates the CK0403-induced
growth inhibition of SKBR-3 cells
As shown in Fig. 1,
among the cell lines tested in the present study, the
HER2-overexpressing SKBR-3 cell line was most sensitive to CK0403
treatment. Since SKBR-3 cells overexpress the HER2 receptor, SKBR-3
cells were selected to evaluate the combined effects of CK0403 and
herceptin. At the tested doses, the growth inhibitory effects of
CK0403 on SKBR-3 cells were found to be potentiated by the
herceptin added at 1/20 of the concentration of CK0403 (Table II).
| Table IIPotentiation of the growth inhibition
activity of CK0403 by herceptin in SKBR-3 human breast cancer
cellsa. |
Table II
Potentiation of the growth inhibition
activity of CK0403 by herceptin in SKBR-3 human breast cancer
cellsa.
| Concentration of
CK0403 (µM) | Percentage of cell
survival (%)
|
|---|
| CK0403 | Herceptinb | CK0403 +
Herceptin |
|---|
| 0.03125 | 50.3 | 75.9 | 45.3 |
| 0.125 | 39.5 | 91.6 | 26.1 |
| 1 | 19.8 | 69.2 | 4.3 |
Discussion
Structurally, CK0402, CK0403 and amsacrine are
9-aminoacridine analogues. Amsacrine was the first clinically
successful synthetic DNA-intercalating agent (8). Amsacrine and its analogs contain a
DNA-binding domain (acridine moiety) and a topoisomerase II-binding
domain (aniline moiety). Amsacrine itself, distinct from
anthracyclines, is not affected by transport-mediated MDR; in
addition, its topoisomerase II-binding domain can be modified to
overcome the observed atypical MDR (10). Thus, structural modifications of
amsacrine analogs may provide effective second-line therapeutics
for tumors that have developed resistance to doxorubicin and
etoposide. Certain amsacrine analogues have been found to be potent
against the human breast carcinoma T-47D cell line and showed
activity in phase II clinical studies on breast cancer (17,18).
In spite of these successes, continuous efforts are made to develop
novel 9-aminoacridine analogues with enhanced activity against
breast cancer in pre-clinical studies (13,14,19–21).
A previous study by our group showed that CK0402
possesses activity against a broad spectrum of cancer types,
including a panel of established human breast cancer cell lines
(15). CK0403 is an analogue of
CK0402 and was found to inhibit the topoisomerase-IIα catalyzed
decatenation reaction ~10 times more effectively than amsacrine and
its CK0402 analogue (14). The
present study, determined that CK0403 is more potent than its
CK0402 analogue against several breast cancer cell lines,
particularly to the HER2-overexpressing SKBR-3 cell line and the
triple-negative MDA-MB-231 cell line. Of note, the non-cancerous
MCF10A cell line was the least sensitive cell line among the cell
lines tested, indicating that CK0403 may selectively kill malignant
cells over non-malignant cells. Based on the growth-inhibitory
effects induced by CK0403 on the panel of cell lines examined in
the present study, CK0403 appears to be more effective against
malignant ER(−) cells than against ER(+) cells.
The tumor microenvironment has a crucial role in the
chemoresistance of tumor cells. A hypoxic environment in solid
tumors is common, which results in increased anaerobic glycolysis,
new blood vessel formation, genetic instability and a decreased
responsiveness to radio- as well as chemotherapy (22,23).
Therefore, tumor hypoxia represents a challenge for
chemotherapeutic agents. Autophagy is induced by a variety of
metabolic stresses, including hypoxia and oxidative stress.
Increasing evidence has shown that autophagy contributes to the
resistance to anti-neoplastic agents, including topoisomerase II
inhibitors (24,25). A previous study by our group showed
that autophagy as well as apoptosis can be induced by CK0402 in
SKBR-3 cells (15); however, the
role of autophagy in CK0402-induced cytotoxic effects has remained
elusive. The results of the present study indicated that the growth
inhibitory activity of CK0403 was reduced, although not distinctly,
under hypoxic conditions. Whether autophagy contributes to the
cellular resistance to cell death induced by CK0403 will be the
subject of further study. The LC50 value was in the
nanomolar range under hypoxic as well as normoxic conditions,
demonstrating that CK0403 effectively inhibited the growth of
SKBR-3 cells under either condition.
The present study further demonstrated that CK0403
induced the cleavage of Atg5 in SKBR-3 cells more effectively than
CK0402, amsacrine, doxorubicin and camptothecin at the doses used.
The process of autophagosome formation requires the covalent
conjugation of Atg12 and Atg5 in a ubiquitylation-like process
(26). However, the non-conjugated
forms of Atg12 and Atg5 are involved in the induction of apoptosis;
in apoptotic cells, Atg5 is cleaved by calpains and then
translocates to the mitochondria, resulting in the release of
cytochrome C by interacting with B-cell lymphoma 2 family proteins
(16,26).
The present and a previous study by our group showed
that CK0402 and CK0403 induced G2/M arrest in breast
cancer cells, which is a common cellular response to treatment with
DNA-damaging agents (15). In
addition, the sensitivity of various breast cancer cell lines to
CK0403 was consistent with that to CK0402. A previous study by our
group determined that apoptosis was the type of cell death induced
by CK0402 in breast cancer cells. In a similar fashion, CK0403
effectively induced apoptosis in SKBR-3 cells, as demonstrated in
the present study. Based on the above results and the structural
similarity between the two compounds, it is concluded that CK0403
inhibited breast cancer cell growth through similar mechanisms to
those of CK0402, but with higher potency. Collectively, our the
results of the present study supported that CK0403 is superior to
CK0402 as a potential treatment option for ER-negative and
HER2-overexpressing breast cancers; furthermore, the growth
inhibitory effects on HER2-overexpressing breast cancer cells were
potentiated by the addition of herceptin.
Acknowledgments
The present study was supported by a grant from the
Susan G. Komen Breast Cancer Foundation (no. BCTR0707876) and the
Tobacco settlement fund from the Pennsylvania Department of
Health.
References
|
1
|
Cancer Facts & Figures 2014. American
Cancer Society; Atlanta: 2014
|
|
2
|
Gianni L and Valagussa P: Anthracyclines
and early breast cancer: The end of an era? J Clin Oncol.
27:1155–1157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Shaughnessy J: Liposomal anthracyclines
for breast cancer: Overview. Oncologist. 8(Suppl 2): S1–S2. 2003.
View Article : Google Scholar
|
|
4
|
Qian BJ, Yan F, Li N, Liu QL, Lin YH, Liu
CM, Luo YP, Guo F and Li HZ: MTDH/AEG-1-based DNA vaccine
suppresses lung metastasis and enhances chemosensitivity to
doxorubicin in breast cancer. Cancer. Immunol Immunother.
60:883–893. 2011. View Article : Google Scholar
|
|
5
|
Weiss RB: The anthracyclines: Will we ever
find a better doxorubicin? Semin Oncol. 19:670–686. 1992.PubMed/NCBI
|
|
6
|
Carvalho FS, Burgeiro A, Garcia R, Moreno
AJ, Carvalho RA and Oliveira PJ: Doxorubicin-induced
cardiotoxicity: From bioenergetic failure and cell death to
cardiomyopathy. Med Res Rev. 34:106–135. 2014. View Article : Google Scholar
|
|
7
|
Ng R, Better N and Green MD: Anticancer
agents and cardiotoxicity. Semin Oncol. 33:2–14. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Denny WA: DNA-intercalating ligands as
anti-cancer drugs: Prospects for future design. Anticancer Drug
Des. 4:241–263. 1989.PubMed/NCBI
|
|
9
|
Minotti G, Menna P, Salvatorelli E, Cairo
G and Gianni L: Anthracyclines: Molecular advances and
pharmacologic developments in antitumor activity and
cardiotoxicity. Pharmacol Rev. 56:185–229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baguley BC, Holdaway KM and Fray LM:
Design of DNA intercalators to overcome topoisomerase II-mediated
multidrug resistance. J Natl Cancer Inst. 82:398–402. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Denny WA: Acridine derivatives as
chemotherapeutic agents. Curr Med Chem. 9:1655–1665. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nelson EM, Tewey KM and Liu LF: Mechanism
of antitumor drug action: Poisoning of mammalian DNA topoisomerase
II on DNA by 4′-(9-acridinylamino)-methanesulfon-m-anisidide. Proc
Natl Acad Sci USA. 81:1361–1365. 1984. View Article : Google Scholar
|
|
13
|
Chen KM, Sun YW, Tang YW, Sun ZY and Kwon
CH: Synthesis and antitumor activity of sulfur-containing
9-anilinoacridines. Mol Pharm. 2:118–128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Park SK, Kang H and Kwon CH:
Caspase-dependent cell death mediates potent cytotoxicity of
sulfide derivatives of 9-anilino-acridine. Anticancer Drugs.
19:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun YW, Niu TK, Yang JM, Kwon CH, Chen KY
and Chen KM: Potentiation of growth inhibition activity of
2-({4-[4-(acridin-9-ylamino)phenylthio]phenyl}(2-hydroxyethyl)
amino) ethan-1-ol (CK0402) by Herceptin in SKBR-3 human breast
cancer cells. Exp Ther Med. 1:513–518. 2010.PubMed/NCBI
|
|
16
|
Yousefi S, Perozzo R, Schmid I, Ziemiecki
A, Schaffner T, Scapozza L, Brunner T and Simon HU:
Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis.
Nat Cell Biol. 8:1124–1132. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baguley BC, Denny WA, Atwell GJ, Finlay
GJ, Rewcastle GW, Twigden SJ and Wilson WR: Synthesis, antitumor
activity and DNA binding properties of a new derivative of
amsacrine, N-5-dimethyl-9-[(2-methoxy-4-methylsulfonylamino)
phenylamino]-4-acridinecarboxamide. Cancer Res. 44:3245–3251.
1984.PubMed/NCBI
|
|
18
|
Fyfe D, Price C, Langley RE, Pagonis C,
Houghton J, Osborne L, Woll PJ, Gardner C, Baguley BC and
Carmichael J: Cancer Research Campaing Phase I/II Trials Committee:
A phase I trial of amsalog (CI-921) administered by intravenous
infusion using a 5-day schedule. Cancer Chemother Pharmacol.
47:333–337. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bacherikov VA, Chou TC, Dong HJ, Zhang X,
Chen CH, Lin YW, Tsai TJ, Lee RZ, Liu LF and Su TL: Potent
antitumor 9-anilinoacridines bearing an alkylating N-mustard
residue on the anilino ring: Synthesis and biological activity.
Bioorg Med Chem. 13:3993–4006. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kapuriya N, Kapuriya K, Zhang X, Chou TC,
Kakadiya R, Wu YT, Tsai TH, Chen YT, Lee TC, Shah A, et al:
Synthesis and biological activity of stable and potent antitumor
agents, aniline nitrogen mustards linked to 9-anilinoacridines via
a urea linkage. Bioorg Med Chem. 16:5413–5423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su TL, Lin YW, Chou TC, Zhang X,
Bacherikov VA, Chen CH, Liu LF and Tsai TJ: Potent antitumor
9-anilinoacridines and acridines bearing an alkylating N-mustard
residue on the acridine chromophore: Synthesis and biological
activity. J Med Chem. 49:3710–3718. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chun YS, Adusumilli PS and Fong Y:
Employing tumor hypoxia for oncolytic therapy in breast cancer. J
Mammary Gland Biol Neoplasia. 10:311–318. 2005. View Article : Google Scholar
|
|
23
|
Adamski JK, Estlin EJ and Makin GW: The
cellular adaptations to hypoxia as novel therapeutic targets in
childhood cancer. Cancer Treat Rev. 34:231–246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tamura N, Hirano K, Kishino K, Hashimoto
K, Amano O, Shimada J and Sakagami H: Analysis of type of cell
death induced by topoisomerase inhibitor SN-38 in human oral
squamous cell carcinoma cell lines. Anticancer Res. 32:4823–4832.
2012.PubMed/NCBI
|
|
26
|
Rubinstein AD and Kimchi A: Life in the
balance-a mechanistic view of the crosstalk between autophagy and
apoptosis. J Cell Sci. 125:5259–5268. 2012. View Article : Google Scholar
|