Introduction
Cardiac hypertrophy is associated with several forms
of heart disease, including ischemic disease, hypertensive heart
disease and valvular stenosis, and is a major risk factor for the
development of heart failure and subsequent mortality (1). Despite advances in the treatment of
heart failure, it remains one of the leading causes of death in
industrialized countries (2).
Therefore, elucidation of the mechanisms underlying the progression
of cardiac hypertrophy to heart failure is important to develop
effective therapeutic strategies for the treatment of heart
failure.
Netrin-1 is a laminin-associated protein and is
identified as a neuronal guidance cue, directing axons to its
targets during the development of the nervous system (3). Netrin-1 mediates its functions
through stimulation of the deleted in colorectal cancer (DCC)
family receptors, DCC and neogenin, and the UNC5A, UNC5B, UNC5C and
UNC5D UNC5 family receptors (4).
In addition to its primary function in neuronal development, the
expression of netrin-1 outside the nervous system inhibitsthe
migration of leukocytes in vitro and in vivo, and
attenuates inflammation-mediated tissue injury (5–7). The
administration of netrin-1 to mice suppresses infiltration and
inflammation in sepsis, AKI, acute lung injury, peritoneal
inflammation and whole body hypoxia (8–13).
Previous studies have demonstrated that netrin-1 prevents
ischemia/reperfusion-induced myocardial infarction (14–16)
and a study by Joseph and Quan indicated netrin-1 as a new
therapeutic target in cardiovascular disease (17), however, the role of netrin-1 in
cardiac hypertrophy has not been investigated.
In the present study, the role of netrin-1 in the
development of cardiac hypertrophy and heart failure was
investigated. The expression levels of netrin-1 were decreased in
the TAC mice and in neonatal rat cardiomyocytes in response to ET-1
stimulation. In addition, the loss of netrin-1 aggravated foetal
gene expressions induced by ET-1 stimulation. By contrast, this
increase of gene expression was suppressed by the overexpression of
netrin-1, and netrin-1 eliminated ventricular remodeling, cardiac
dysfunction and DNA damage during pressure overload. Furthermore,
analysis of the signaling events indicated that the
netrin-1-mediated protection against cardiac hypertrophy was
attributed to interruption of the activation of the
mitogen-activated protein kinase (MAPK) kinase (MEK) kinase-1
(K1)-dependent MEK-extracellular signal-regulated protein kinase
1/2 (ERK1/2) and c-Jun N-terminal kinase 1/2 (JNK1/2) signaling
pathways.
Materials and methods
Pressure overload models
A total of 18 male 3 month-old wild-type C57 mice
weighing 15–20 g and maintained at 25–30°C were obtained from
Tongji Medical College (Wuhan, China). The mice were anaesthetized
by intraperitoneal injection with a mixture of ketamine (80
mg/kg/h) and xylazine (8 mg/kg/h; Sigma-Aldrich, St. Louis, MO,
USA), intubated, and artificially ventilated, as previously
described (18). Pressure overload
was then induced by performing thoracic transverse aortic
constriction (TAC). A standard lead II electrocardiogram was used
for recordings throughout the experiment, and the adequacy of
anaesthesia was monitored from the disappearance of the pedal
withdrawal reflex. A number of animals were administered via tail
vein injection with recombinant netrin-1 (eBioscience, Houston, TX,
USA) at a dose of 5 µg/mouse every 3 days. Sham surgery
involved opening the chest without performing thoracic transverse
aortic constriction. The cardiac function was evaluated 4 weeks
after TAC or sham-surgery by transthoracic echocardiography, using
an FFsonic 8900 (Fukuda Denshi Co., Tokyo, Japan) equipped with a
13 MHz phased-array transducer, under anaesthesia with
intraperitoneal pentobarbital sodium (35 mg/kg; Sigma-Aldrich). The
adequacy of anaesthesia was monitored at all times by assessment of
skeletal muscle tone, respiratory rate and rhythm, and response to
tail pinch. Left-ventricular (LV) fractional shortening (LVFS) was
calculated as [LV end-diastolic diameter (LVEDD) – LV end-systolic
diameter) / LVEDD] x 100(%). The mice in the TAC and sham groups
were then sacrificed by intraperitoneal injection of ketamine (1
g/kg) and xylazine (100 mg/kg), and their hearts were rapidly
excised. The mRNA levels of Mouse A- and B-type natriuretic
peptides (ANP and BNP) were determined by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). The
present study was approved by the Ethics Committee of the People's
Hospital of Gansu (Lanzhou, China).
Cultured neonatal rat cardiomyocytes
Hearts were collected from 1–2-day-old male Sprague
Dawley neonatal rat pups weighing 2–3 g (Tongji Medical College),
promptly following euthanasia by decapitation. Primary cultures of
neonatal rat cardiomyocytes were performed, as described previously
(19,20). Following serum starvation, the
neonatal rat cardiomyocytes were stimulated with endothelin-1
(ET-1; Sigma-Aldrich), and samples were collected to examine the
expression levels of netrin-1 by western blot analysis and the mRNA
expression of ANP by qPCR. Netrin-1 siRNA was purchased from Thermo
Scientific Dharmacon and was used to transfect the cardiomyocytes
using GenomOne-Neo (Ishihara Sangyo Kaisha, Osaka, Japan),
according to the manufacturer's instructions. The activities of the
ET-1-inducible ANP and BNP promoters were evaluated using a
luciferase(luc) reporter gene assay with human (h)ANP/luc and
BNP/luc.
RT-qPCR
Total RNA was extracted from the cultured cells and
tissues using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA) and reverse transcribed into cDNA using the
PrimeScript RT reagent kit (Takara Biotechnology, Dalian, China),
according to the manufacturer's instructions. The mRNA levels of
target genes were quantified using SYBR Green Master mix (Takara
Biotechnology) using an ABI Prism 7900 Sequence Detector system
(Applied Biosystems, Foster City, CA). Each reaction was performed
in duplicate, and changes in the relative gene expression
normalized to levels of 18s RNA were determined using the relative
threshold cycle method (21).
Immunohistochemistry
The myocardial sections (5-µm sections of
cryostat frozen tissue) from the mice were stained with mouse
monoclonal anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) antibody
(1:200; clone N45.1; eBioscience) to evaluate the degree of DNA
damage in the heart. The staining was visualized by treatment with
a solution of 3,3′-diaminobenzidine (Dako Cytomation Liquid DAB
Substrate Chromogen System; Dako Japan, Tokyo, Japan) for 40 sec at
4°C. The 8-OHdG-positive area was measured (five random fields to
yield ~400 cardiomyo-cytes) using Image J software version 1.46
(National Institutes of Health, Bethesda, MD, USA). An Olympus
optical microscope was used (Axio Lab.A1 MAT; Olympus, Tokyo,
Japan).
Western blotting
The protein levels of netrin-1, phosphorylated
(p-)ERK1/2, p-MEK1/2, p-JNK1/2 and p-P38 were determined by western
blot analysis. The protein extracted from the cells or tissues was
separated on 10% SDS-polyacrylamide electrophoresis gels
(Sigma-Aldrich) and transferred onto nitrocellulose membranes
(Pierce Biotechnology, Rockford, IL, USA). Following being blocked
with 5% non-fat milk in Tris-buffered saline for 3 h, the membranes
were incubated with the indicated primary antibodies (0.2
µg/ml; rabbit polyclonal ERK1/2 antibody, cat. no.
16443-1-AP, Proteintech, Chicago, IL, USA; rabbit anti-phospho-JNK
1/2, cat. no. Rs-1640R, Sigma-Aldrich; rabbit monoclonal MEK1
antibody, cat. no. AJ1468a, Abgent, San Diego, CA, USA; rabbit
polyclonal p38 antibody, cat. no. NB100-56665, Novus Biologicals,
Littleton, CO, USA) at 4°C overnight, followed by incubation with
horseradish peroxidase-conjugated goat anti-rabbit IgG secondary
antibody (1:5,000; eBioscience) for 3 h. All the lanes were probed
for β-actin as loading controls and the proteins were detected
using an enhanced chemiluminescence detection kit (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Differences between the groups were evaluated using
one-way analysis of variance with Bonferroni's post-hoc test.
Survival curves, following TAC, were generated using the
Kaplan-Meier method and were then compared using the log-rank test.
P<0.05 was considered to indicate a statistically significant
difference. The statistical analyses were performed using the
standard statistical software program JMP version 8 (SAS Institute
Inc., Cary, NC, USA).
Results
Expression of netrin-1 decreases in
murine hearts following TAC
To investigate the expression of netrin-1 during LV
remodeling, the gene and protein levels of netrin-1 was
investigated in heart samples from mice following pressure
overload, generated by TAC. It was found that the expression of
netrin-1 was markedly decreased following TAC (Fig. 1A and B). Furthermore, the
expression of netrin-1 was examined in neonatal rat cardiomyocytes
following ET-1 stimulation. Similar to the observations in the
heart samples from the TAC mice, the expression of netrin-1 was
suppressed following ET-1 stimulation (Fig. 1C).
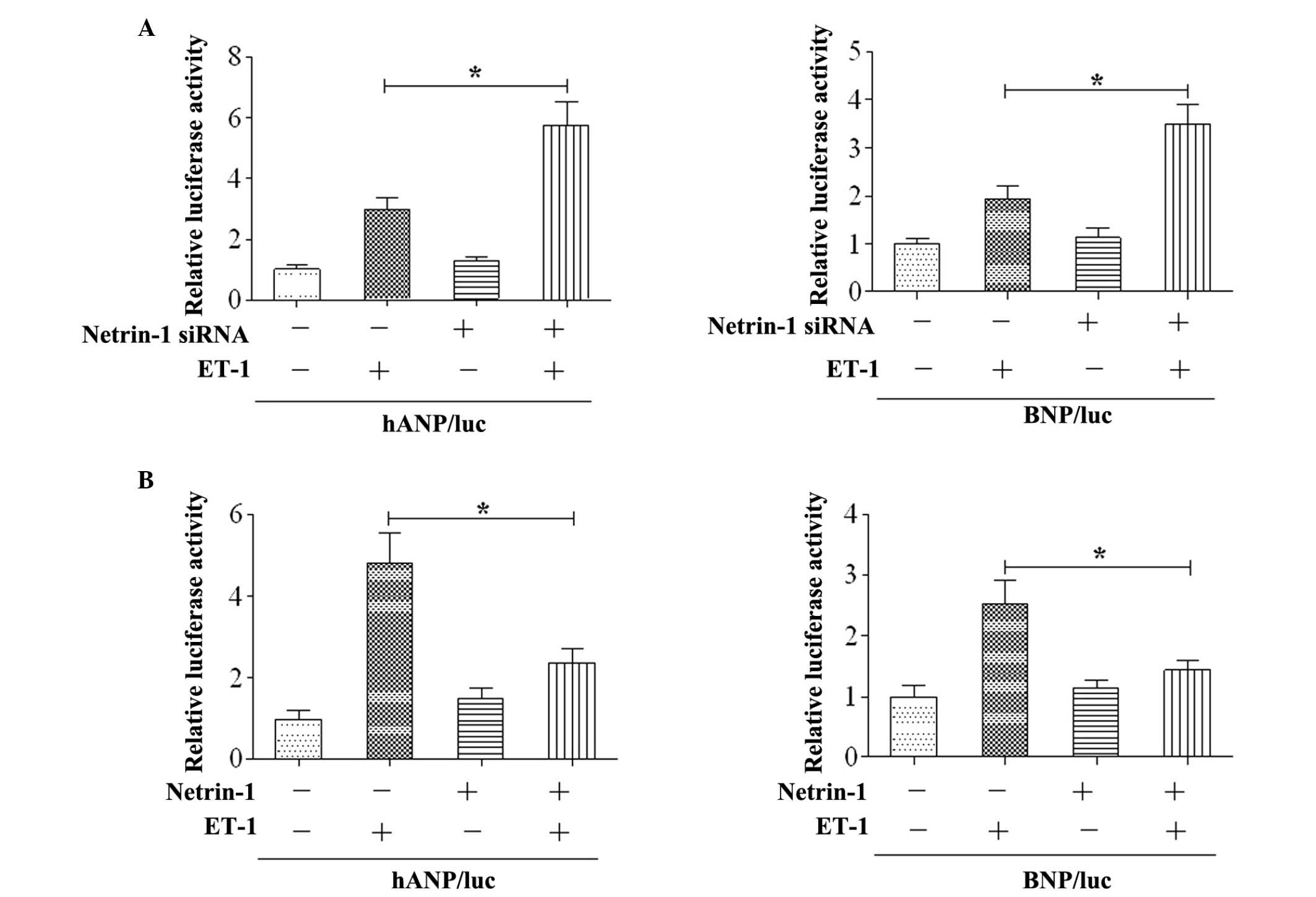
Netrin-1 suppresses the expression of
cardiac foetal gene
To determine the role of netrin-1 on cardiac
hypertrophy, the expression of foetal cardiac gene was examined
in vitro. The results revealed that the activities of the
ANP and BNP promoters were increased by ET-1 stimulation in the
cardiomyocytes, and co-transfection with netrin-1 siRNA enhanced
the activities of the ANP and BNP promoters (Fig. 2A). Subsequently, netrin-1 was
co-transfected with the ANP/luc or BNP/luc constructs. The results
demonstrated that overexpression of netrin-1 significantly
attenuated the activities of the ANP and BNP promoters following
ET-1 stimulation (Fig. 2B).
Netrin-1 attenuates the development of
cardiac hypertrophy and heart failure
To examine the role of netrin-1 in the development
of cardiac hypertrophy and heart failure in vivo, mice were
subjected to either TAC or sham surgery. At 4 weeks following the
TAC surgery, the increase in the weight of the hearts was
significantly lower in Uthe mice treated with netrin-1 (Fig. 3A). The expression levels of ANP,
BNP and β-MHC were significantly upregulated in the TAC group
compared with the sham group, and this increase was markedly
attenuated in the TAC mice treated with netrin-1 (Fig. 3B). Furthermore, systolic
dysfunction and left ventricular dilation subsequent to TAC were
attenuated in mice administered with netrin-1 compared with the
control (Fig. 3C). The survival
rate following TAC was significantly higher in mice treated with
netrin-1 compared with those without netrin-1 treatment (Fig. 3D).
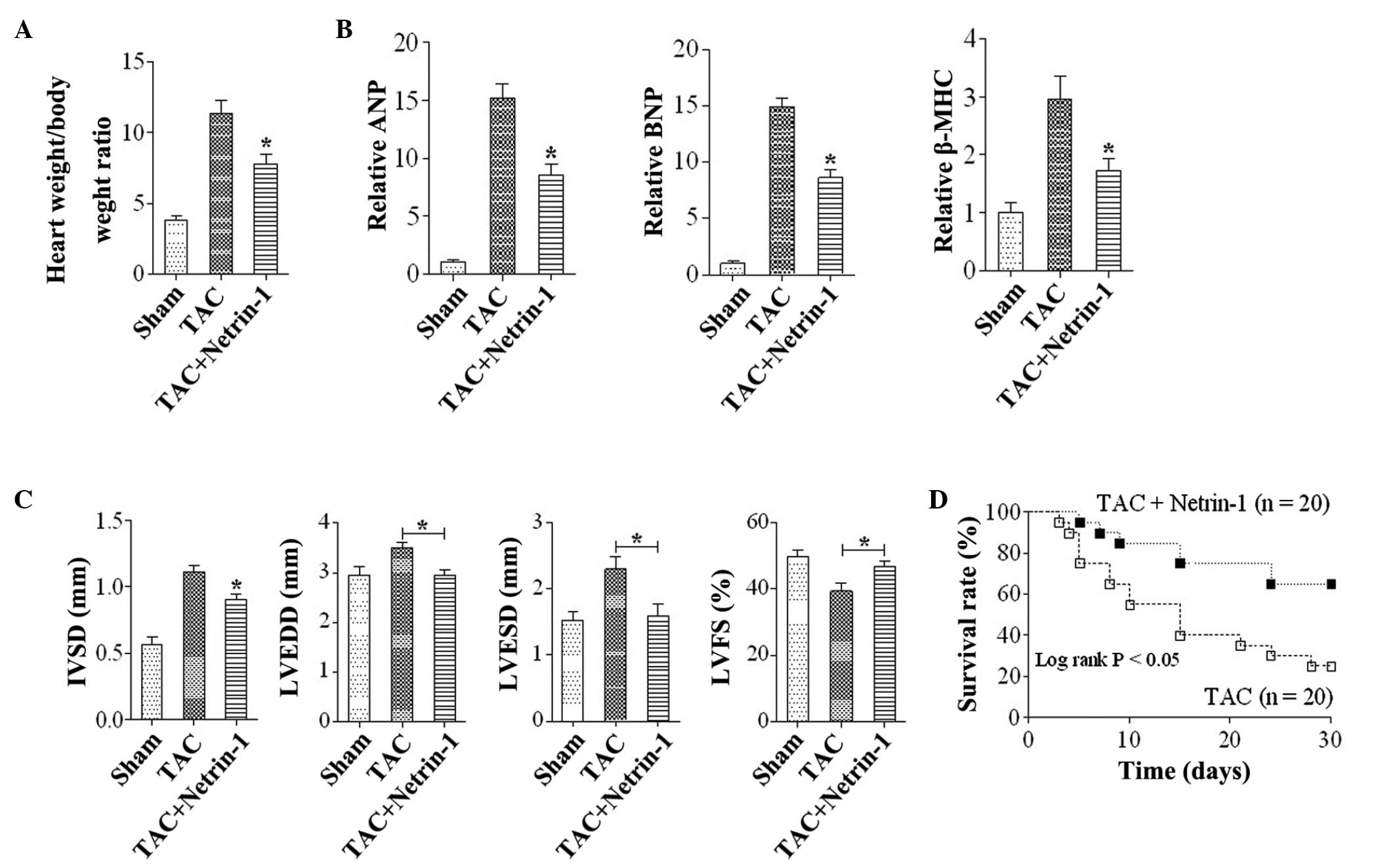 | Figure 3Netrin-1 attenuates the development of
cardiac hypertrophy and heart failing. (A) Heart weight: body
weight ratios in the TAC and sham group mice. (B) Quantitative
analysis of the gene expression levels of ANP, BNP and β-MHC in
mice from the Sham, TAC and TAC+Netrin-1 groups.
*P<0.05, vs. Sham and TAC (n=6). (C) Data from the
echocardiographic measurements in mice from the Sham, TAC and TAC +
Netrin-1 groups. *P<0.05, vs. Sham and TAC for IVSD;
*P<0.05, vs TAC group for LVEDD, LVESD and %LVFS
(n=6). Data are expressed as the mean ±standard error of the mean.
(D) Survival curves of mice in the TAC and TAC + netrin-1 groups.
IVSD, interventricular wall thickness; LVEDD, left-ventricular
end-diastolic dimension; LVESD, left-ventricular end-systolic
dimension;%LVFS, left-ventricular fractional shortening; TAC,
transverse aortic constriction; Sham, no TAC; MHC, major
histocompatibility complex; Sham, no TAC; ANP, A-type natriuretic
peptide; BNP, B-type natriuretic peptide. |
To investigate the role of netrin-1 in protecting
cardiomyocytes from DNA damage in cardiac hypertrophy,
immunohistochemical staining of the hearts from the TAC group were
performed using anti-8-OHdG antibody. The mice in the sham group
did not exhibit 8-OHdG-positive cardiomyocytes. In the TAC mice,
the expression of 8-OHdG was significantly increased, whereas the
induction of 8-OHdG was suppressed in the netrin-1-treated mice
(Fig. 4).
Netrin-1 inhibits the pressure
overload-mediated MEK-ERK1/2 and JNK1/2 signaling pathways
The results of the present study demonstrated that
netrin-1 has a protective role in cardiac hypertrophy. However, the
molecular mechanisms through which netrin-1 treatment affects the
hypertrophic responses on stress stimuli remain to be elucidated.
To determine these possible mechanisms, the present study
investigated the expression and activity of the MAPK signaling
molecules, MEK1/2, ERK1/2, JNK1/2 and P38, as the MAPK pathway is
known to be involved in pathological cardiac hypertrophy (22). The expression levels of p-MEK1/2,
ERK1/2, and JNK1/2 were markedly increased in the TAC group
compared with the sham group, and this increase was markedly
attenuated in the TAC mice treated with netrin-1 (Fig. 5).
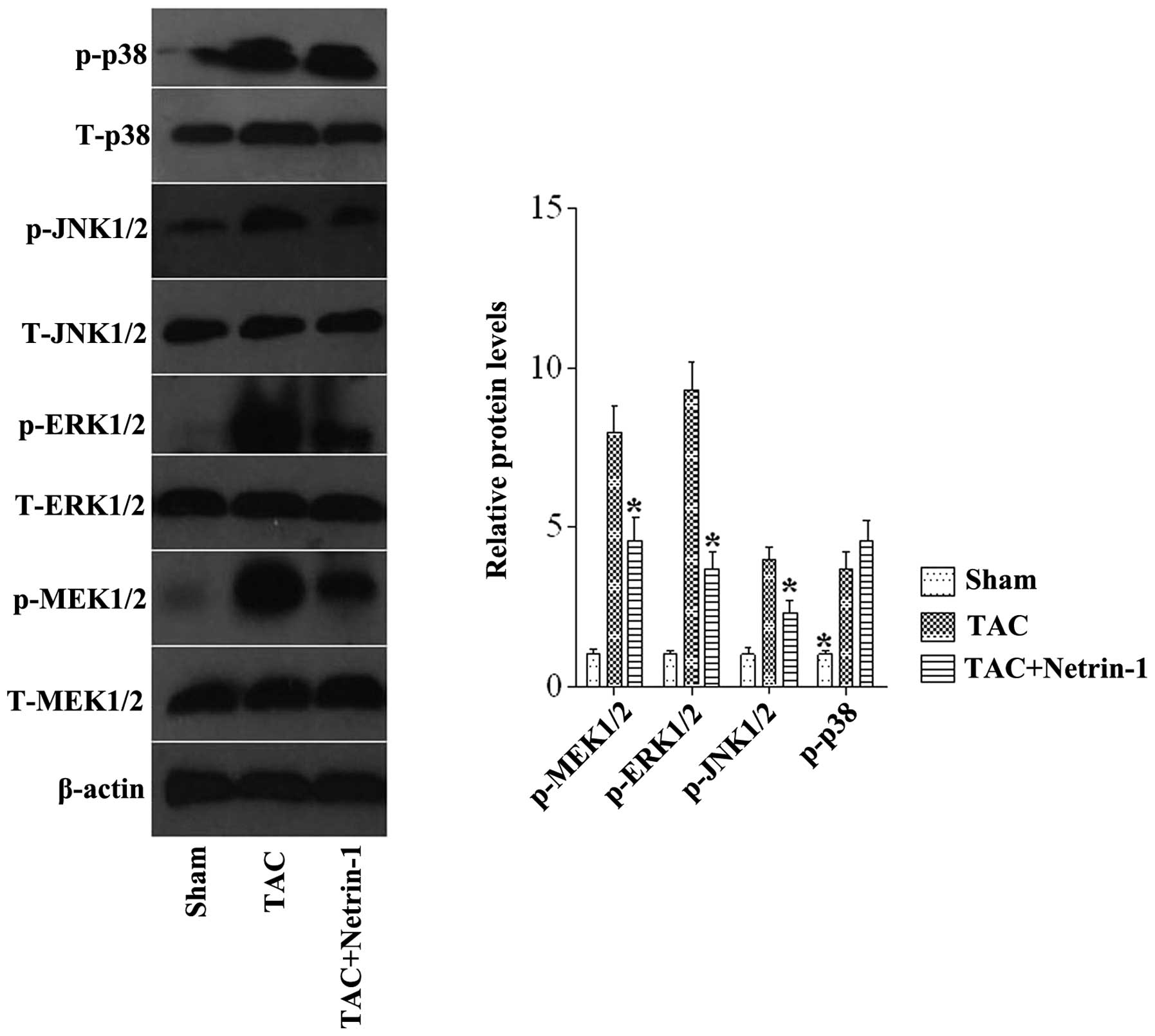 | Figure 5Representative western blots and
quantitative results of the phosphorylated and total protein levels
of MEK1/2, ERK1/2, JNK1/2 and P38 in mice from the Sham, TAC, and
TAC + netrin-1 groups. *P<0.05 vs. all other groups
(n=6). Data are expressed as the mean ±standard error of the mean.
TAC, transverse aortic constriction; Sham, no TAC; p-,
phosphorylated; MEK, mitogen-activated protein kinase kinase; ERK,
extracellular-regulated protein kinase; JNK, c-Jun N-terminal
kinase. |
Discussion
The present study demonstrated the importance of
netrin-1 in the development of cardiac hypertrophy and heart
failure. The results revealed that the expression of netrin-1 was
decreased in TAC mice and neonatal rat cardiomyocytes in response
to ET-1 stimulation. The results also demonstrated that the loss of
netrin-1 aggravated the expression of foetal genes induced by the
ET-1 stimulation. By contrast, this increase in gene expression
levels was suppressed by treatment with netrin-1, and netrin-1
eliminated ventricular remodeling, cardiac dysfunction and DNA
damage during pressure overload. Furthermore, the analyses of the
signaling events indicated that the netrin-1-mediated protection
against cardiac hypertrophy was attributed to interrupting the
MEKK1-dependent MEK-ERK1/2 and JNK1/2 signaling pathways.
Netrin-1 is a laminin-associated molecule and is
secreted at the spinal cord midline, where it is involved in
guiding vertebrate commissural axons. A previous study reported
that netrin-1 is expressed by the vascular endothelium,
particularly in post capillary venules (23). The present study demonstrated that
the expression of foetal genes induced by ET-1 was inhibited by the
overexpression of netrin-1. In addition, the pressure
overload-induced expression of foetal genes is attenuated following
treatment of netrin-1 compared with control. Pressure
overload-induced oxidative stress contributes to cardiac DNA damage
and DNA repair/synthesis in failing hearts with systolic
dysfunction (24). Therefore, DNA
damage is considered to be a key pathogenic factor in ventricular
dysfunction (25). A previous
study demonstrated the involvement of netrin-1 in protecting
against DNA-damage (26). In the
present study, 8-OHdG induction following TAC was significantly
attenuated in the netrin-1-treated mice compared with control.
Taken together, these findings indicated that netrin-1 may prevent
DNA damage during pressure overload, as observed in the decrease in
cardiac dysfunction in mice treated with netrin-1. A decrease in
netrin-1 induced by hypertrophic stimulation may cause DNA damage,
increasing the severity of cardiac hypertrophy, affecting the
expression of foetal genes and causing cardiac dysfunction.
The mechanism underlying the antihypertrophic effect
of netrin-1 remains to be fully elucidated. Considerable evidence
exists to indicate that activation of the MAPK signaling pathway
contributes to the pathogenesis of cardiac hypertrophy (27). MAPK signaling is mediated through
three-kinase cascades of associated protein isoforms, beginning
with an upstream MAPKKK (MEKK) and leading sequentially to MEK and
effector MAPK. MAPK pathways have been implicated in a wide array
of cellular responses to environmental stimuli (28,29).
Extensive evidence has documented the involvement of subdivisions
of the ERK, JNK and p38 MAPKs in specific aspects of cardiac
remodeling (30,31). To clarify the molecular mechanisms
involved in the suppressive effect of netrin-1 in cardiac
hypertrophy, the present study evaluated the activation status of
the MAPK pathway in hypertrophic models. The results demonstrated
that the activation of MEK1/2, ERK1/2 and JNK1/2 was significantly
enhanced in response to chronic pressure overload, and this
upregulation was markedly inhibited by netrin-1. However, netrin-1
administration did not effect the protein expression of P38 in the
hypertrophic models. Therefore, it is conceivable that netrin-1
exerted its antihypertrophic effects through inhibition of
MEK-ERK1/2 and JNK1/2 signaling.
In conclusion, the present study provided evidence
to support the hypothesis that netrin-1 protects against pressure
overload-induced cardiac hypertrophy and heart failure through
negative regulation of the MEKK1-dependent MEK-ERK1/2 and JNK1/2
signaling pathways. These findings suggest that netrin-1 may be a
novel therapeutic target for the prevention of pathological cardiac
hypertrophy.
References
|
1
|
McMurray JJ and Pfeffer MA: Heart failure.
Lancet. 365:1877–1889. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roger VL, Weston SA, Redfield MM, et al:
Trends in heart failure incidence and survival in a community-based
population. JAMA. 292:344–350. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tessier-Lavigne M and Goodman CS: The
molecular biology of axon guidance. Science. 274:1123–1133. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barallobre MJ, Pascual M, Del RJ and
Soriano E: The Netrin family of guidance factors: emphasis on
Netrin-1 signalling. Brain Res Brain Res Rev. 49:22–47. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tadagavadi RK, Wang W and Ramesh G:
Netrin-1 regulates Th1/Th2/Th17 cytokine production and
inflammation through UNC5B receptor and protects kidney against
ischemia-reperfusion injury. J Immunol. 185:3750–3758. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosenberger P, Schwab JM, Mirakaj V, et
al: Hypoxia-inducible factor-dependent induction of netrin-1
dampens inflammation caused by hypoxia. Nat Immunol. 10:195–202.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang W, Reeves WB, Pays L, Mehlen P and
Ramesh G: Netrin-1 overexpression protects kidney from ischemia
reperfusion injury by suppressing apoptosis. Am J Pathol.
175:1010–1018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ly NP, Komatsuzaki K, Fraser IP, et al:
Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc
Natl Acad Sci USA. 102:14729–14734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mirakaj V, Thix CA, Laucher S, et al:
Netrin-1 dampens pulmonary inflammation during acute lung injury.
Am J Respir Crit Care Med. 181:815–824. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mirakaj V, Gatidou D, Potzsch C, Konig K
and Rosenberger P: Netrin-1 signaling dampens inflammatory
peritonitis. J Immunol. 186:549–555. 2011. View Article : Google Scholar
|
|
11
|
Rosenberger P, Schwab JM, Mirakaj V, et
al: Hypoxia-inducible factor-dependent induction of netrin-1
dampens inflammation caused by hypoxia. Nat Immunol. 10:195–202.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grenz A, Dalton JH, Bauerle JD, et al:
Partial netrin-1 deficiency aggravates acute kidney injury. PLoS
One. 6:e148122011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aherne CM, Collins CB, Masterson JC, et
al: Neuronal guidance molecule netrin-1 attenuates inflammatory
cell trafficking during acute experimental colitis. Gut.
61:695–705. 2012. View Article : Google Scholar :
|
|
14
|
Zhang J and Cai H: Netrin-1 prevents
ischemia/reperfusion-induced myocardial infarction via a
DCC/ERK1/2/eNOS s1177/NO/DCC feed-forward mechanism. J Mol Cell
Cardiol. 48:1060–1070. 2010. View Article : Google Scholar :
|
|
15
|
Durrani S, Haider KH, Ahmed RP, Jiang S
and Ashraf M: Cytoprotective and proangiogenic activity of ex-vivo
netrin-1 transgene overexpression protects the heart against
ischemia/reperfusion injury. Stem Cells Dev. 21:1769–1778. 2012.
View Article : Google Scholar :
|
|
16
|
Ahmed RP, Haider KH, Shujia J, Afzal MR
and Ashraf M: Sonic Hedgehog gene delivery to the rodent heart
promotes angiogenesis via iNOS/netrin-1/PKC pathway. PLoS One.
5:e85762010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Joseph BB and Quan PD: The neuroimmune
guidance cue netrin-1: a new therapeutic target in cardiovascular
disease. Am J Cardiovasc Dis. 3:129–134. 2013.PubMed/NCBI
|
|
18
|
Shishido T, Woo CH, Ding B, et al: Effects
of MEK5/ERK5 association on small ubiquitin-related modification of
ERK5: implications for diabetic ventricular dysfunction after
myocardial infarction. Circ Res. 102:1416–1425. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Funayama A, Shishido T, Netsu S, et al:
Cardiac nuclear high mobility group box 1 prevents the development
of cardiac hypertrophy and heart failure. Cardiovasc Res.
99:657–664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Le NT, Takei Y, Shishido T, et al: p90RSK
targets the ERK5-CHIP ubiquitin E3 ligase activity in diabetic
hearts and promotes cardiac apoptosis and dysfunction. Circ Res.
110:536–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jones ME, Mayne GC, Wang T, Watson DI and
Hussey DJ: A fixed-point algorithm for estimating amplification
efficiency from a polymerase chain reaction dilution series. BMC
Bioinformatics. 15:3722014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Tang QZ, Liu C, et al: Cellular
FLICE-inhibitory protein protects against cardiac remodeling
induced by angiotensin II in mice. Hypertension. 56:1109–1117.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ly NP, Komatsuzaki K, Fraser IP, et al:
Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc
Natl Acad Sci USA. 102:14729–14734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siggens L, Figg N, Bennett M and Foo R:
Nutrient deprivation regulates DNA damage repair in cardiomyocytes
via loss of the base-excision repair enzyme OGG1. FASEB J.
26:2117–2124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Suzuki S, Shishido T, Ishino M, et al:
8-Hydroxy-2′-deoxyguanosine is a prognostic mediator for cardiac
event. Eur J Clin Invest. 41:759–766. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Ozaki T, Shamim HM, et al: A newly
identified dependence receptor UNC5H4 is induced during DNA
damage-mediated apoptosis and transcriptional target of tumor
suppressor p53. Biochem Biophys Res Commun. 370:594–598. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Van Berlo JH, Maillet M and Molkentin JD:
Signaling effectors underlying pathologic growth and remodeling of
the heart. J Clin Invest. 123:37–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Garrington TP and Johnson GL: Organization
and regulation of mitogen-activated protein kinase signaling
pathways. Curr Opin Cell Biol. 11:211–218. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kuida K and Boucher DM: Functions of MAP
kinases: insights from gene-targeting studies. J Biochem.
135:653–656. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ravingerova T, Barancik M and Strniskova
M: Mitogen-activated protein kinases: a new therapeutic target in
cardiac pathology. Mol Cell Biochem. 247:127–138. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Y: Mitogen-activated protein kinases
in heart development and diseases. Circulation. 116:1413–1423.
2007. View Article : Google Scholar : PubMed/NCBI
|