Introduction
Occult macular dystrophy (OMD) is an inherited
macular disease characterized by progressive visual decline with
the absence of visible fundus abnormalities (1). The condition was first recognized in
1989 as an autosomal dominant trait, although sporadic cases have
also been reported (1-3). In patients with OMD, the full-field
electroretinogram (ERG) is normal; however, the focal macular ERG
and multifocal ERG (mfERG) of the macular area are affected,
revealing localized dysfunction of the photoreceptors (1,4,5).
Despite normal fundus appearance, spectral domain
optical coherence tomography (SD-OCT) reveals various degrees of
macular changes in OMD patients. Typical OCT features of OMD
patients are a disruption of the inner segment/outer segment
(IS/OS) junction and the disappearance of the cone outer segment
tip (COST) line (6). Subtle
changes of normal macular reflectance may also be noted in infrared
reflectance images of the fundus (7).
Mutations in the retinitis pigmentosa 1-like 1
(RP1L1) gene have been identified in four Japanese families with
OMD (8). To date, the association
between OMD and RP1L1 mutations has mostly been reported in Asian
subjects (7-10). Only two studies on Caucasian
subjects with OMD are available, of which only one included an
analysis for RP1L1 gene mutations (11,12).
The present study was the first to report on an Italian family with
typical OMD and a RP1L1 mutation.
Materials and methods
Patient examination and clinical data
collection
In 2013, a Caucasian family comprising several
members diagnosed with OMD was assessed. In accordance with the
Declaration of Helsinki, all patients provided written informed
consent to full ophthalmological examination and genetic testing
for OMD mutations. All subjects were evaluated by expert
ophthalmologists at the Opthalmology Associates in Padova (Italy).
Each subject underwent complete ophthalmological assessment,
including best-corrected visual acuity (BCVA), slit-lamp
examination, IOP measurement and ophthalmos-copy. Color vision
testing was performed using Ishihara pseudochromatic plates.
High-resolution macular scans were obtained by SD-OCT (3D-OCT 2000;
Topcon, GB Ltd., Newbury, UK). The medical records of the subjects
were also thoroughly analyzed.
Genetic testing
Genetic testing was performed at the Research and
Innovation Laboratories Srl (Padova, Italy). DNA extraction was
performed from peripheral blood using the Qiagen Biorobot blood
extraction kit (Qiagen, Hilden, Germany) according to maufacturer's
instructions. 100 ng DNA were amplified by standard polymerase
chain reaction (PCR) procedures with the PCR mixture containing 2.5
µl 10X concentrated PCR buffer (Solis Biodyne, Tartu,
Estonia), 0.7 µl 50 mM MgCl2 (Solis Biodyne),
0.75 µl 10 mM deoxyribo-nucleotide triphosphates (Solis
Biodyne), 2.5 µl S solution (Solis Biodyne), 0.3 µl
100 µM forward and reverse primer (IDT, Coralville, IA, USA;
primer sequences are listed in Table
I) and 0.5 µl 5 U/µl Hot Start DNA Polymerase
(Solis Biodyne). Termocycling consisted in 1 cycle of enzyme
activation (15 min at 95°C) followed by 35 cycles of DNA
amplification (45 sec at 95°C, 45 sec at 59°C and 1 min at 72°C).
PCR products were then separated by agarose gel electrophoresis
(1.5% agarose gel in tris-borate-ethylenediaminetetraacetate;
Sigma-Aldrich, St. Louis, MO, USA) and purified by Invisorb spin
columns (Invitek, Hayward, CA, USA). PCR-purified products were
re-amplified with terminated nuclotides employing Big Dye
Terminator v3.1 (Applied Biosystems; Thermo Fisher Scientific,
Waltham, MA, USA). Sequencing analysis was performed using an ABI
Prism 3100 Avant automated sequencer (Thermo Fisher Scientific)
equipped with 36-cm capillary array filled with POP6 polymer
(Thermo Fisher Scientific). Electropherograms were analyzed using
Sequencing Analysis software (version 3.5; Applied Biosystems).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Primer name | Primer sequence | Mutations |
|---|
| RP1L1 1199 Fw |
GGGCTCTCATAAGTTCTTGAATCAG | Ser1199Cys |
| RP1L1 1199 Rv |
GCAGATCATGAGGGCGCT |
| RP1L1 960 Fw |
AAGGAGAAGCAGTGCCAGCC | Trp960Arg |
| RP1L1 960 Rv |
TGCTGTCCCGCCTGAGC |
| RP1L1 45 Fw |
AAGAGACAGGAAATGCCAATCC | Arg45Trp |
| RP1L1 45 Rv |
TCTTATCAGAGCAGAGGTAGCAGC |
| RP1L1 2311 Fw |
CTTCACTGGCCCCCTGCT | Gln2311Pro,
Gln2311Glu |
| RP1L1 2311 Rv |
GCCCTCAGGTCAGTCTAGGAGAT |
| RP1L1 1425 Fw |
CGTGTGCTCTTGGCCCAT | Asp1425Glu,
Asp1425His |
| RP1L1 1425 Rv |
TGCAGTTAGAGGAAGTTAAAGAAGGG |
Results
Ophthalmological assessment
As illustrated in Fig.
1, the study included six members of an Italian family (four
males and two females), among which three presented with OMD
(subjects, II 1, III 1 and III 2).
The main patient (subject II 1) was a 67-year-old
male. The BCVA was ʻcounting fingersʼ in both eyes (OU) without any
detectable alteration of the anterior and posterior segment of the
eye. The patient's medical records revealed a progressive visual
decline beginning at age 59. The results of the slit-lamp
examination as well as intraocular pressure and fundus appearance
were normal. The results of the color vision test was also normal
until the BCVA reached 0.3. Automated perimetry revealed a central
relative scotoma in OU. Full-field ERG was within normal limits,
whereas multi-focal ERG showed a reduction of the amplitudes in the
central region. Clinical and instrumental findings were matched the
diagnosis of OMD.
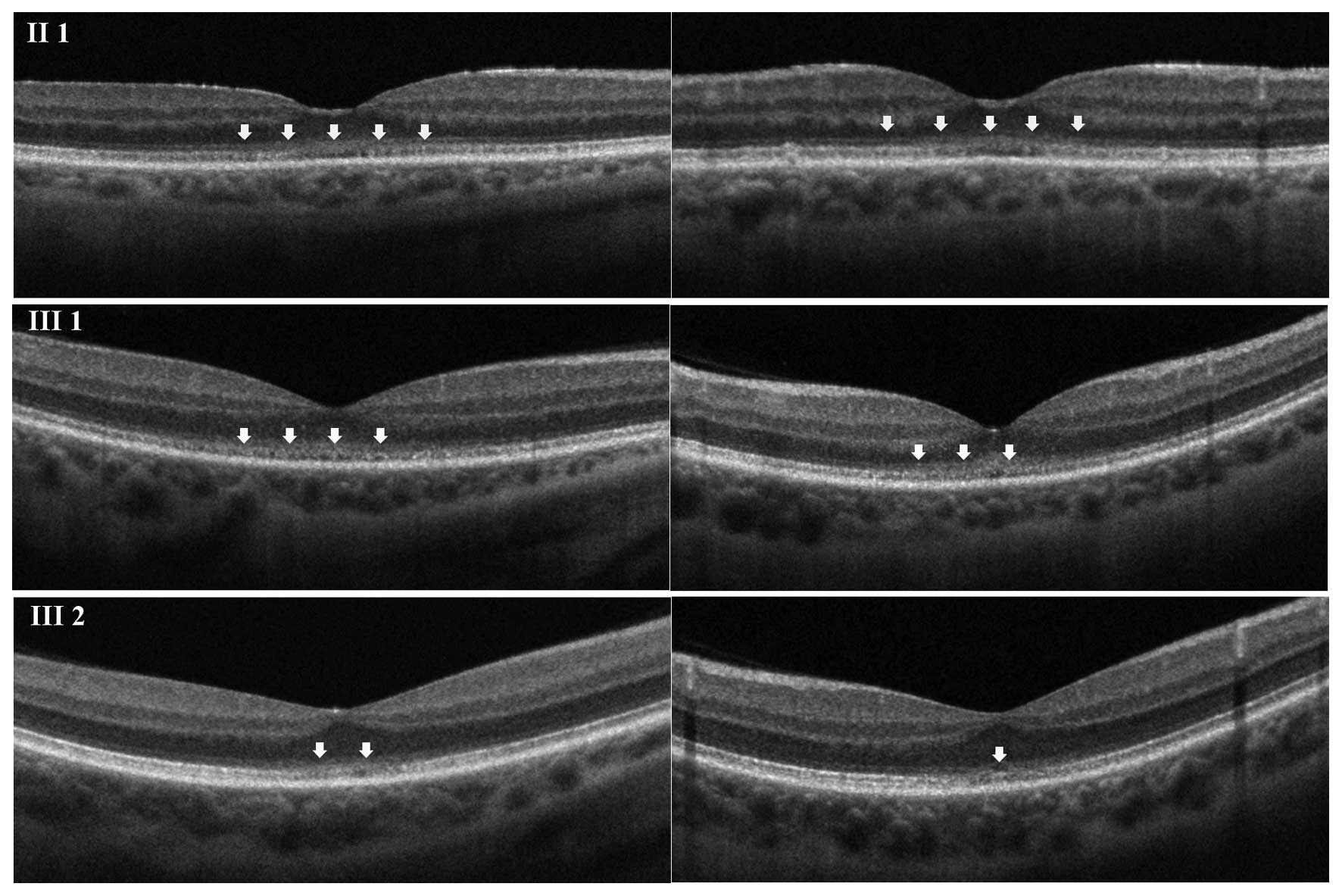
SD-OCT was then performed; macular scans showed that
in the foveal region, the external limiting membrane (ELM) and the
inner segment-outer segment (IS/OS) line were not sharply
identifiable from each other. In addition, the cone outer segment
tip (COST) line was disrupted (Fig.
2). A mild hyperreflectivity of the internal limiting membrane
was also present, and central retinal thickness was reduced.
The second patient (subject III 1) was a 38-year-old
male. The BCVA was 0.2 in OU, and no ocular abnormalities were
detected by slit-lamp and fundus examination. The patient's medical
records reported progressive visual decline, associated with
photophobia, starting at age 15. Color vision testing showed a
red-green defect and automated perimetry revealed a central
relative scotoma in OU. SD-OCT results regarding the ELM, the IS/OS
line and the COST line were similar to those observed in subject II
1; however, the affected area was smaller (Fig. 2).
Subject III 2 was a 32 year-old male. The subject's
BCVA was 0.6 in OU, with no evidence of any changes in ocular
morphology. Color vision testing showed normal results. The patient
presented with a slow, progressive decline of visual acuity since
age 28. The SD-OCT findings were comparable to those in subjects II
1 and III 1, although the area involved was minimal (Fig. 2).
In addition, clinical assessment of three clinically
asymptomatic siblings of subject II 1 (II 2, II 3 and II 4, a 51
year-old male, a 56 year-old female and a 66 year-old female,
respectively) was performed (Fig.
1). All three subjects had a BCVA of 1.0 in OU and normal
results of the ophthalmological examination. SD-OCT revealed that
the retinal layers were normal.
Analysis of RP1L1 mutations
Subject II 1 was screened for the following targeted
mutations in the RP1L1 gene: Ser1199Cys, Trp960Arg, Arg45Trp,
Gln2311Pro, Gln2311Glu, Asp1425His and Asp1425Glu. Of these, the
Arg45Trp mutation, which was reported by Akahori et al
(8) to be associated with
autosomal-dominant OMD, was identified in subject II (Fig. 3A). As Arg45Trp was previously found
to be correlated with occult macular dystrophy, subjects III 1, III
2, II 2, II 3 and II 4 were also screened for this specific
mutation. As expected due to their clinical diagnosis, subjects III
1 and III 2 were also carriers of the Arg45Trp mutation (Fig. 3B and C). While subjects II 2 and II
4 were also carriers of the Arg45Trp mutation, they displayed no
clinical symptoms of OMD (Fig. 3D and
F). The Arg45Trp mutation was not present in subject II3
(Fig. 3E). Therefore, in the study
population, the Arg45Trp mutation was present in 3/5 subjects
(60%).
Discussion
The prevalence of occult macular dystrophy (OMD)
appears to be elevated Asian populations, as the majority of
available studies have been performed in Japanese or Korean
populations (1,4-10).
Only two studies on Caucasian subjects with OMD are available, of
which only one included an analysis for RP1L1 gene mutations
(11,12).
OMD is an autosomal inherited condition, although
sporadic cases have also been reported. RP1L1 was identified as a
gene responsible for the disease in 2010 (8). RP1L1 is located on chromosome 8p and
originates from the common ancestor gene retinitis pigmentosa 1
(RP1) on chromosome 8. RP1L1 shares 35% amino acid identity with
RP1, a gene responsible for 51% of autosomal dominant retinitis
pigmen-tosa cases worldwide (8,11).
RP1 is responsible for 5-10% of autosomal dominant retinitis
pigmentosa cases worldwide (11).
Although RP1L1 shares 35% amino acid identity with RP1, RP1L1
mutations are not associated with retinitis pigmentosa.
Immunohistochemical studies on RP1L1 in retinal
sections of cynomolgus monkeys showed that the gene is expressed in
rods as well as cones (8).
Although the function of RP1L1 remains to be fully elucidated, it
probably cooperates with RP1 to assemble and stabilize the
microtubules of the photoreceptor axonemes (13).
While the link between OMD and RP1L1 mutations has
been reported in Asian and north European pedigrees, the present
study was the first to demonstrate that an RP1L1 mutation is likely
to be associated with OMD in an Italian family. The substitution
mutation p.Arg45Trp was identified in a patient who met the
diagnostic criteria for OMD, as well as in the two symptomatic male
offspring of the patient and also in two clinically asymptomatic
siblings of the patient. Tsunoda et al (10), who reported on a Japanese family
carrying the p.Arg45Trp mutation, found that the age of onset of
OMD ranged from 6-50 years. It is often difficult to establish the
true age of onset of the disease, as certain patients with OMD have
normal visual acuity and no subjective visual disturbance until the
disease progresses to a more advanced stage (10). In the pedigree assessed in the
present study, the age of first presentation of visual symptoms
ranged from 15 years for subject III 1 to 59 years of age for
subject II 1. The onset of progressive BCVA reduction reported by
Tsunoda et al (10) ranged
from 6-50 years of age. Similarly, in the present study, the onset
of progressive BCVA reduction varied. Similarly, in the present
study, the rate of the progression of the visual decline varied.
Subject II 1 became legally blind over a period of eight years,
while subject III 1 showed a slower reduction of BCVA, which
decreased to 0.2 over a period of 23 years. BCVA of the third
proband decreased to 0.6 over three years; however, the time of
observation was too short to predict the rate of progression. OMD
has been reported to be a slowly progressing disease, while the
visual function usually became stationary when the final visual
acuity reached a value of 0.1–0.2 (8,10).
However, the BCVA of Subject II 1 deteriorated to counting fingers
in OU.
The diagnosis of OMD is challenging, as no
detectable fundus abnormalities are present at advanced stages.
Despite normal fundus appearance, SD-OCT is able to identify a
number of alterations in the outer retinal layers in the majority
of OMD patients. Tomographic features of OMD include central
retinal thinning, blurring of the ELM and IS/OS band as well as
disruption of the COST line, whereas the RPE band is always
preserved (6-7,10).
All of these characteristics were observed in the OMD patients of
the present study. It has also been reported that the degree of
these alterations worsens over time and correlates with visual
impairment. In spite of the small number of patients included in
the present study, it was indicated that a better visual acuity is
correlated with less extensive macular alterations.
Thorough analysis of SD-OCT scans of asymptomatic
subjects II 2, II 3 and II 4 revealed that no major or minimal
alterations of the outer retinal layers were present. Tsunoda et
al (10) reported that even
though certain OMD patients showed a good BCVA or did not complain
about any visual symptoms, the mfERG revealed the presence of a
macular dysfunction.
In conclusion, the present study was the first to
demonstrate the occurrence of the p.Arg45Trp mutation in the RP1L1
gene in an Italian pedigree including OMD patients. The RP1L1
Arg45Trp mutation, at least in the cohort of the present study,
appeared to have incomplete penetrance. Davidson et al
(12) suggested the Arg45Trp
mutation as a risk factor for OMD rather than a causative mutation.
However, the reason for the incomplete penetrance displayed by
individuals possessing the OMD-associated RP1L1 variant p.Arg45Trp
remains elusive. A digenic or oligogenic model involving other
genes requires further investigation. In the present study, the
first of South-European origin, a disease penetrance of 60% was
presented; this is higher than that observed by Davidson et
al (12) (38%), and more
comparable with a study on Asian subjects by Akahori et al
(8) (identified 85% penetrance).
In addition Davidson et al (12) concluded that the Arg45Trp mutation
is a risk factor, not a causative mutation, for OMD, based on the
observation that none of their patients had familial history of
autosomal-dominant maculopathy. In the current study, a Caucasian
father and his two sons are described who presented with OMD, thus
supporting the role of the Arg45TRp mutation in the cause of the
disease in non-Asian subjects. Of note, as the age of onset for OMD
is variable, clinical diagnosis of OMD is challenging and healthy
carriers of the Arg45Trp mutation should be considered to be at
risk, and periodic clinical screening for OMD is recommended.
Based on previous studies and the observations of
the present study, it is indicated that screening for mutations in
the RP1L1 gene associated with progressive reduction of BCVA and
typical OCT per se may be sufficient to confirm the diagnosis of
OMD. Under these circumstances, mfERG is not necessary (as in
subjects III 1 and III 2). However, when visual symptoms or fundus
abnormalities are absent, but genetic testing is positive, mfERG
may be the only test with the capability of early detection of an
impending macular dysfunction.
References
|
1
|
Miyake Y, Ichikawa K, Shiose Y and Kawase
Y: Hereditary macular dystrophy without visible fundus abnormality.
Am J Ophthalmol. 108:292–299. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mattews GP, Sandberg MA and Berson EL:
Foveal cone electroretinograms in patients with central visual loss
of unexplained etiology. Arch Ophthalmol. 110:1568–1570. 1992.
View Article : Google Scholar
|
|
3
|
Lyons JS: Non familial occult macular
dystrophy. Doc Ophthalmol. 111:49–56. 2005. View Article : Google Scholar
|
|
4
|
Miyake Y, Horiguchi M, Tomita N, Kondo M,
Tanikawa A, Takahashi H, Suzuki S and Terasaki H: Occult macular
dystrophy. Am J Ophthalmol. 122:644–653. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Piao CH, Kondo M, Tanikawa A, Terasaki H
and Miyake Y: Multifocal electroretinogram in occult macular
dystrophy. Invest Ophthalmol Vis Sci. 41:513–517. 2000.PubMed/NCBI
|
|
6
|
Park SJ, Woo SJ, Park KH, Hwang JM and
Chung H: Morphologic photoreceptor abnormality in occult macular
dystrophy on spectral-domain optical coherence tomography. Invest
Ophthalmol Vis Sci. 51:3673–3679. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahn SJ, Ahn J, Park KH and Woo SJ:
Multimodal imaging of occult macular dystrophy. JAMA Ophthalmol.
131:880–890. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Akahori M, Tsunoda K, Miyake Y, Fukuda Y,
Ishiura H, Tsuji S, Usui T, Hatase T, Nakamura M, Ohde H, et al:
Dominant mutations in RP1L1 are responsible for occult macular
dystrophy. Am J Hum Genet. 87:424–429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kabuto T and Takahashi H, Goto-Fukuura Y,
Igarashi T, Akahori M, Kameya S, Iwata T, Mizota A, Yamaki K,
Miyake Y and Takahashi H: A new mutation in RP1L1 gene in a patient
with occult macular dystrophy associated with a depolarizing
pattern of focal macular electroretinograms. Mol Vis. 18:1031–1039.
2012.
|
|
10
|
Tsunoda K, Usui T, Hatase T, Yamai S,
Fujinami K, Hanazono G, Shinoda K, Ohde H, Akahori M, Iwata T and
Miyake Y: Clinical characteristics of occult macular dystrophy in a
family with mutation of RP1L1 gene. Retina. 32:1135–1147. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jacobson SG, Cideciyan AV, Iannaccone A,
Weleber RG, Fishman GA, Maguire AM, Affatigato LM, Bennett J,
Pierce EA, Danciger M, et al: Disease expression of RP1 mutations
causing autosomal dominant retinitis pigmentosa. Invest Ophthalmol
Vis Sci. 41:1898–1908. 2000.PubMed/NCBI
|
|
12
|
Davidson AE, Sergouniotis PI, Mackay DS,
Wright GA, Waseem NH, Michaelides M, Holder GE, Robson AG, Moore
AT, Plagnol V and Webster AR: RP1L1 variants are associated with a
spectrum of inherited retinal diseases including retinitis
pigmentosa and occult macular dystrophy. Hum Mutat. 34:506–514.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamashita T, Liu J, Gao J, LeNoue S, Wang
C, Kaminoh J, Bowne SJ, Sullivan LS, Daiger SP, Zhang K, et al:
Essential and synergistic roles of RP1 and RP1L1 in rod
photoreceptor axoneme and retinitis pigmentosa. J Neurosci.
29:9748–9760. 2009. View Article : Google Scholar : PubMed/NCBI
|