Introduction
Drowning is a major, but often neglected, public
health problem (1). Drowning is
the third leading cause of accidental mortality, with >388,000
mortalities per year worldwide (2). Water inhalation can induce acute lung
injury (ALI) and acute respiratory distress syndrome by disturbing
the barrier function of alveolar epithelium, leading to lung edema
and inflammatory reactions (3–5).
Previous research has demonstrated that intracellular calcium
(Ca2+) oscillations are vital in ALI, as they lead to
reduced integrity of the blood-air barrier (6,7),
increased NF-κB activation and lung inflammation (7). It was also indicated that
intracellular Ca2+ oscillations are dependent on
extracellular Ca2+ (7),
and an increased infiltration coefficient can be prevented in
low-Ca2+ lung perfusate (8). Therefore, modulating calcium
signaling may provide beneficial effects in cases of ALI induced by
seawater aspiration.
Transient receptor potential-vanilloid (TRPV) is a
family of plasma membrane ion channels consisting of seven members
(TRPV1-7) (9). They are a notable
receptor family that respond to a wide variety of endogenous and
exogenous stimuli, including temperature (10,11),
proinflammatory mediators (11–13),
pH (11,14), stretch (15) and osmolality (11,16,17).
Two members of the TRPV family (TRPV1 and 4) have been identified
to participate in ALI (15,18).
As a cell membrane-bound Ca2+ channel, activation of
TRPV1 is an important factor in intracellular Ca2+
oscillations following exposure to cytokines, abnormal pH and
osmolality (12,18–20).
TRPV4 is also a Ca2+-permeable cation channel gated by a
variety of external factors (17),
including heat (21), membrane
stretch, osmotic changes (22) and
mechanical stimuli (23). Previous
studies have indicated that the activation of TRPV4 and subsequent
Ca2+ entry initiated an acute calcium-dependent
permeability increase following lung injury resulting from
14,15-epoxyeicosatrienoic acid (14,15-EET), 5,6-EET and 8,9-EET
(6) exposure, in addition to
ventilator-induced lung injury (15). These results imply that TRPV is
important during ALI and is a potential therapeutic target for the
treatment of lung injury. Whether seawater exposure induces ALI
through the activation of TRPV remains unknown. The role of the
interaction between hypertonia and high calcium concentration in
seawater instillation-induced ALI required further
investigation.
In the present study, the hypothesis that seawater
instillation induces ALI through the activation of TRPV and
subsequent intracellular calcium oscillations was analyzed and the
interaction between hypertonia and high calcium concentration
during ALI was examined.
Materials and methods
Reagents
The following reagents were used in the current
study: Monoclonal mouse β-actin (1:8,000 dilution; cat. no. A5441;
Sigma-Aldrich, St. Louis, MO, USA); monoclonal rabbit phospho-NF-κB
p65 (Ser536; 1:500 dilution; cat. no. 3033; Cell Signaling
Technology, Inc., Danvers, MA, USA) and monoclonal rabbit NF-κB p65
(1:500 dilution; cat. no. 8242; Cell Signaling Technology, Inc.)
antibodies. Acti-stain 488 phalloidin (Cytoskeleton, Inc., Denver,
USA); enzyme-linked immunosorbent assay (ELISA) kits (R&D
Systems, Inc., Minneapolis, MN, USA); myeloperoxidase (MPO)
activity assay kit (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China); Fluo-3,AM and Pluronic F-127 (Biotium, Inc.,
Hayward, CA, USA); ruthenium red, capsazepine and BAPTA-AM (Abcam);
HC067047 and EGTA (Sigma-Aldrich). Seawater was prepared according
to the major compositions of the East China Sea provided by the
Chinese Ocean Bureau (osmolality, 1,300 mmol/l; pH 8.2; NaCl,
26.518 g/l; MgSO4, 3.305 g/l; MgCl2, 2.447
g/l; CaCl2, 1.141 g/l; KCl, 0.725 g/l;
NaHCO3, 0.202 g/l; NaBr, 0.083 g/l).
Cell culture and treatment
The epithelial cell line A549 (American Type Culture
Collection, Rockville, MD, USA), derived from lung adenocarcinoma
was cultured in RPMI 1640 medium (HyClone Laboratories, Inc.,
Logan, UT, USA) supplemented with 10% fetal bovine serum (Zhejiang
Tianhang Biotechnology Co., Ltd., Huzhou, China) at 37°C and in a
5% CO2 atmosphere. The cells were treated with seawater
by addition to the culture medium at a 25% volume ratio. The cells
and supernatant were harvested 4 h after exposure to seawater.
Single cell [Ca2+]c
measurement
A549 cells grown in culture were exposed to culture
medium containing the Ca2+-sensitive fluorescent
indicator, Fluo-3,AM (5 µM), and a nonionic dispersing
agent, Pluronic F-127 (0.025%), for 30 min at 37°C. Following the
loading period, the medium was replaced, and the cells were
incubated for a further 30 min. Fluorescence intensity, reflecting
the concentration of [Ca2+]c, was recorded by confocal
laser-scanning microscopy (FV10; Olympus, Tokyo, Japan). The
re-addition of calcium was performed by adding CaCl2.
The liquid above the cells was replaced with the seawater with a
normal concentration of CaCl2 (1.141 g/l). Images were
captured for quantification. The groups and their treatments were
as follows: Control group: no extra treatment with the exception of
loading the Fluo-3; seawater group: Following loading with Fluo-3
AM (5 µM), the cells were exposed to seawater challenge at
the predetermined time; BAPTA-AM group: Cells were loaded with
Flou-3 AM (5 µM) and BAPTA-AM (5 µM) 30 min prior to
exposure to seawater challenge at the predetermined time; EGTA
group: Following loading with Fluo-3 AM (5 µM), the cells
were exposed to Ca2+-free seawater with 1 mM EGTA at the
predetermined time; ruthenium red group: Cells were loaded with
Flou-3 AM (5 µM) and ruthenium red (3 µM) 30 min
prior to exposure to seawater challenge at the predetermined time;
HCO67047 group: Cells were loaded with Flou-3 AM (5 µM) and
HCO67047 (1 µM) 30 min prior to exposure to seawater
challenge at the predetermined time; capsazepine group: Cells were
loaded with Flou-3 AM (5 µM) and capsazepine (10 µM)
30 min prior to exposure to seawater challenge at the predetermined
time.
ELISA assay
Levels of TNF-α, IL-1β and IL-6 in lung tissues were
determined using the ELISA kits. Lung tissues were homogenized in
cool phosphate-buffered saline (PBS) at a 1:5 ratio of lung tissue
to PBS. Assays were conducted according to the manufacturer's
instructions. The groups and their treatments were as follows:
Control group: Cells were treated with 25% PBS and 75% RPMI-1640
for 4 h; seawater group: Cells were exposed to 25% seawater and 75%
RPMI-1640 for 4 h; BAPTA-AM group: Cells were loaded with BAPTA-AM
(5 µM) for 30 min and then exposed to 25% seawater and 75%
RPMI-1640 for 4 h; EGTA group: Cells were exposed to 25%
Ca2+-free seawater with 1 mM EGTA and 75% RPMI-1640 for
4 h; seawater + PDTC group: Cells were loaded with PDTC (200
µM) for 30 min and then exposed to 25% seawater and 75%
RPMI-1640 for 4 h; seawater + HCO67047 group: Cells were loaded
with HCO67047 (1 µM) for 30 min and then exposed to 25%
seawater and 75% RPMI-1640 for 4 h; seawater + capsazepine group:
Cells were loaded with capsazepine (10 µM) for 30 min and
then exposed to 25% seawater and 75% RPMI-1640 for 4 h.
Animal procedures
Adult male Sprague-Dawley (SD) rats weighing 180–200
g were purchased from the Laboratory Animal Centre of the Fourth
Military Medical University (Xi'an, China) and housed under a
light/dark cycle of 12/12 h, with standard food and water provided
ad libitum. All procedures used in the present study were
approved by the Animal Care and Use Committee of the Fourth
Military Medical University. Rats were anesthetized with 1.5%
sodium pentobarbital (50 mg/kg; Sigma-Aldrich) followed by
intratracheal administration of seawater (4 ml/kg body weight) into
the lungs within 4 min via a 20-gauge intravenous catheter through
the trachea. The rats were maintained in a supine position with a
30° head-up tilt during the experiments. The rats were euthanized
with a sodium pentobarbital overdose (500 mg/kg) at the
predetermined points of time and then the lungs were harvested for
further experiments. SD rats were randomly assigned into the
following four groups (n=4): Control group: Rats with no
intervention; seawater group: Rats were intratracheally
administered seawater (4 ml/kg body weight) into the lungs within 4
min via a 20-gauge intravenous catheter through the trachea;
Ca2+-free seawater group: Rats were intratracheally
administered Ca2+-free seawater (4 ml/kg body weight)
into the lungs; isotonic seawater group: Rats were intratracheally
administered isotonic seawater with no change in calcium
concentration.
Western analysis
Protein was extracted from the lower right lung by
homogenization and centrifugation at 10,000 × g for 20 min at 4°C.
Proteins were separated by 10% SDS-PAGE (120 v; Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) and were transferred onto
a nitrocellulose membrane (Pall Corp., Washington, NY, USA). The
membranes were blocked with 5% non-fat dry milk in Tris-buffered
saline (TBS) and probed with the antibodies against phospho-NF-κB
p65 (Ser536; dilution, 1:500), NF-κB p65 (dilution, 1:500) and
β-actin (dilution, 1:8,000). Following incubation with the primary
antibody overnight, the membranes were washed with TBS-Tween 20 and
incubated with horseradish peroxidase-conjugated secondary antibody
(dilution, 1:10,000). Target proteins were detected by the enhanced
chemiluminescence detection system (WesternBright ECL-spray ;
Advansta, Inc., Menlo Park, CA, USA). Four samples from each group
were used for densitometry analysis (version 4.6.2; Quantity One
software; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Confocal visualization of F-actin
A549 cells were cultured on coverslips and exposed
to the different treatment conditions, including the control (no
treatment), seawater-treated, seawater + BAPTA-AM and sewater +
capsazepine groups. Following treatments, cells were fixed and
permeabilized at room temperature, washed with PBS and incubated
with 100 nM Acti-stain 488 phalloidin in the dark for 30 min. The
nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole;
Beyotime Institute of Biotechnology, Shanghai, China) staining This
stain was excited using a 340 nm laser and detected by confocal
laser-scanning microscopy. The groups were treated as described
above.
Histopathological evaluation
The lung tissues of the lower lobe of the right lung
harvested from each rat were fixed with 4% paraformaldehyde
(Sigma-Aldrich) for 24 h and embedded in paraffin and
(Sigma-Aldrich) cut into 5-µm sections. The sections were
stained with hematoxylin and eosin (Sigma-Aldrich) prior to
visualization at ×200 magnification under a light microscope (CX41;
Olympus).
MPO activity assay
The extent of neutrophil accumulation in the lung
samples was measured by assaying MPO activity. Following
homogenization and centrifugation (10,000 × g) of these lung
tissues in cool PBS, MPO activity was determined by colorimetric
analysis using a SmartSpec Plus spectrophotometer (Bio-Rad,
Laboratories, Inc.), according to the manufacturer's instructions.
The MPO activity was expressed as U/mg protein.
Evans blue extravasation assessment
Barrier permeability of the lungs was measured by
Evans blue extravasation analysis, and 30 min prior to instillation
of seawater, Evans blue dye (Sigma-Aldrich; 20 mg/kg) was injected
into the rats through the tail vein. Normal saline was injected
into the right ventricle immediately prior to euthanization. Once
clear fluid was effused from the left atrium, the lower lobe of the
right lung was removed. Evans blue dye was extracted from the
tissue by incubation of the lung lobes in formamide (Sigma-Aldrich;
3 ml/100 mg) for 24 h. Total Evans blue (µg/g) was
calculated using spectrophotometry (620 nm; SmartSpec Plus;
Bio-Rad, Laboratories, Inc.).
Bronchoalveolar lavage fluid (BALF)
analysis
Following anesthetization of the rats, the lungs
were lavaged with 1 ml ice-cold PBS three times. The number of
total cells and neutrophils in the BALF was calculated using
Wright's staining (Sigma-Aldrich). The cells in the BALF were
collected by centrifugation at 2,500 × g, stained with Wright's
stain according the manufacturer's instructions and then
neutrophils that were dyed pale purple were counted using a cell
counting plate.
Statistical analysis
All data are expressed as the mean ± standard
deviation. Statistically significant differences between the
different groups were assessed using analysis of variance with a
Bonferroni post-hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Seawater challenge elevated cytosolic
[Ca2+]c by inducing calcium entry from extracellular
medium
It was hypothesized that seawater inhalation leads
to ALI by stimulating Ca2+ entry into the cytosol. In
order to address this issue, a series of experiments were performed
to examine the effects of seawater composed of modified components
on the [Ca2+]c in A549 cells. The effects of seawater
challenge on the [Ca2+]c were recorded using confocal
laser-scanning microscopy to measure the fluorescent
Ca2+-sensitive indicator, Fluo-3,AM, and images were
captured for quantitative analysis. A rise in the fluorescence
intensity indicated an increase in [Ca2+]c. As presented
in Fig. 1A and B, seawater
exposure evoked a rapid [Ca2+]c increase and the
increase reached a maximal value within 30 sec, followed by a
trifling recovery and a sustained plateau. Subsequently, a parallel
experiment in which cells were treated with 5 µM BAPTA-AM (a
selective intracellular Ca2+ chelator) for 40 min prior
to seawater exposure demonstrated that pretreatment with a chelator
completely abolished the elevation of [Ca2+]c induced by
seawater (Fig. 1C).
Next, to clarify the source of Ca2+ ions,
experiments were performed to evaluate whether seawater challenge
elevated [Ca2+]c via release of Ca2+ from
intracellular stores or influx of extracellular Ca2+. As
presented in Fig. 1D, an
extracellular Ca2+ chelator, EGTA, decreased the
increase of [Ca2+]c induced by seawater. The inhibitory
effect was cancelled by re-addition of Ca2+ (Fig. 1D). Thus, it was concluded that the
elevation of [Ca2+]c evoked by seawater exposure was
predominantly accomplished by increasing Ca2+ influx
from extracellular sources. The effects of the different treatments
on [Ca2+]c are summarized in Fig. 1E.
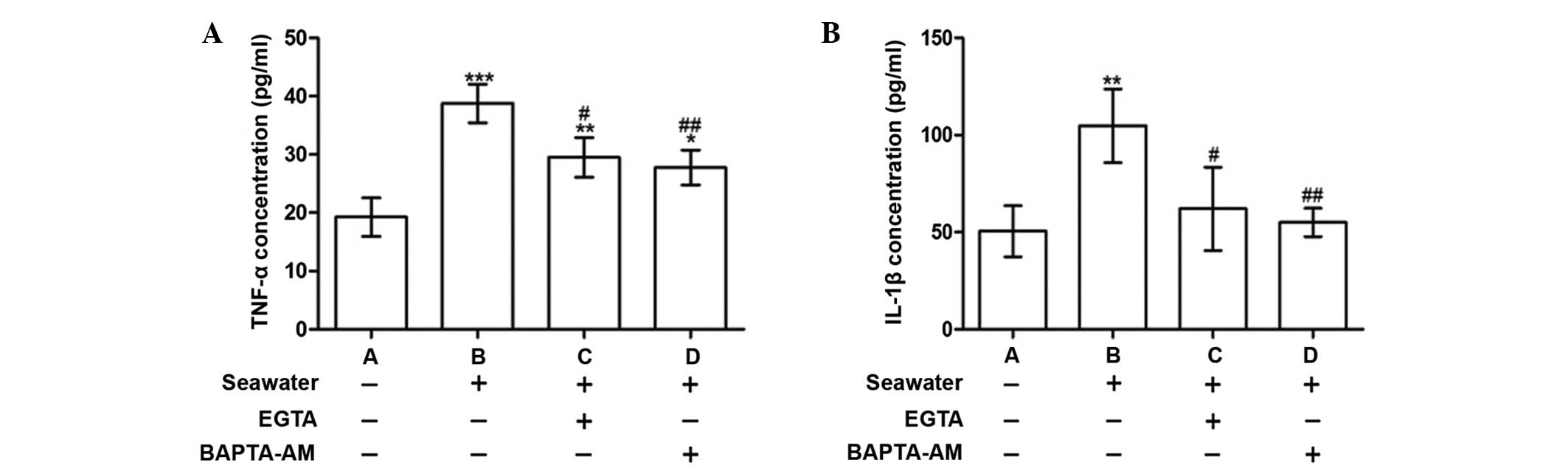
Calcium chelation by EGTA or BAPTA-AM
reduced the release of inflammatory mediators following seawater
exposure
To explore the consequences of [Ca2+]c
elevation, the concentrations of the proinflammatory cytokines,
TNF-α and IL-1β, were measured in the supernatant. Following
seawater exposure, the levels of TNF-α and IL-1β were significantly
increased compared with the control group (P<0.001 and
P<0.01, respectively; Fig. 2).
These alterations were alleviated when cells were treated with
BAPTA-AM or EGTA (Fig. 2).
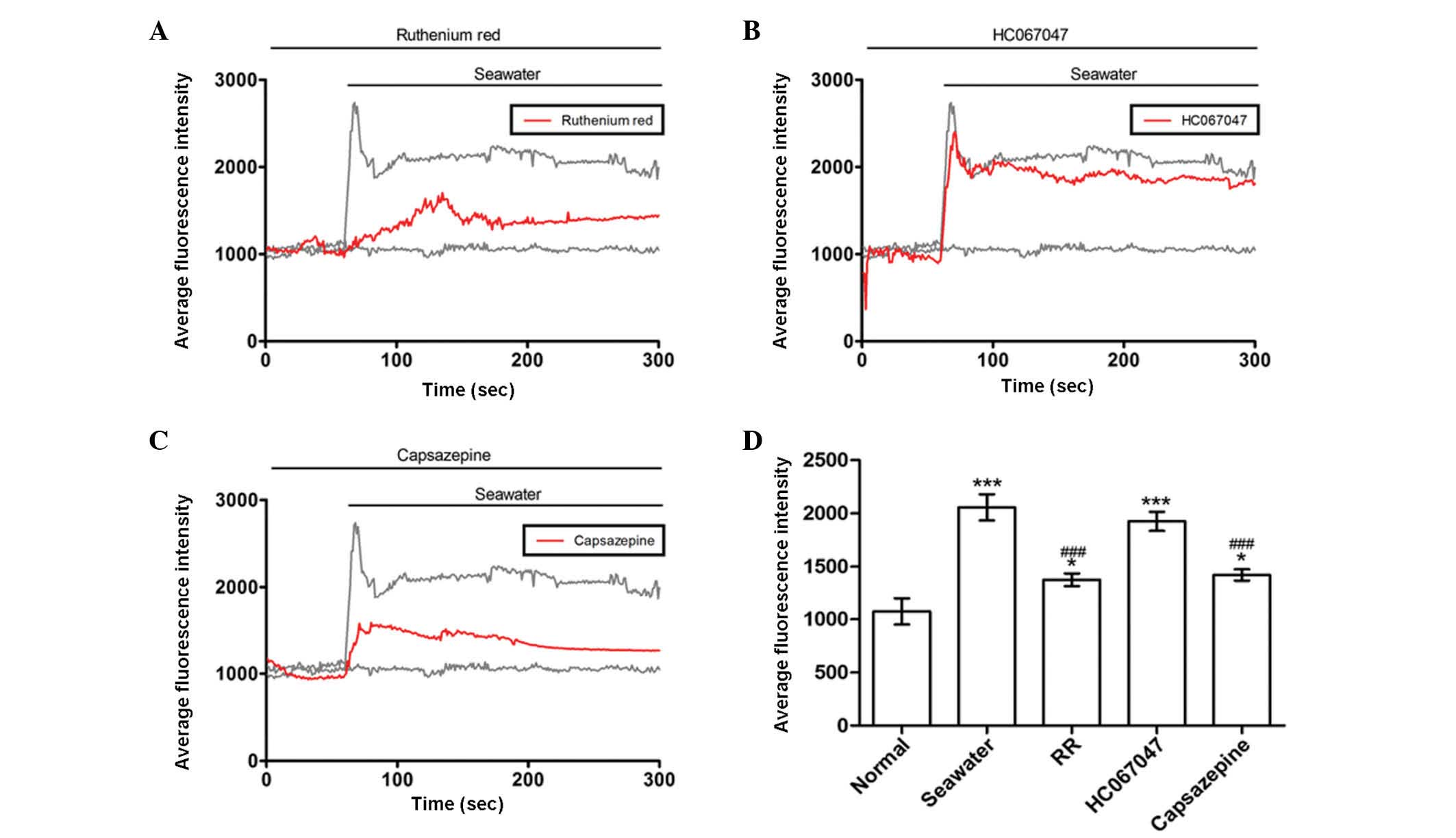
Seawater challenge evoked extracellular
Ca2+ influx by activating TRPV1 channels
It was reported that TRPV4 initiates the acute
calcium-dependent permeability increase during ventilator-induced
lung injury in isolated mouse lungs (6). The current study investigated whether
similar membrane transport pathways were required for seawater
exposure-induced extracellular Ca2+ influx. Subsequent
experiments were then performed to identify the membrane transport
pathway that mediated Ca2+ influx. Cells were treated
with a range of inhibitors of potential Ca2+ entry
channels 30 min prior to seawater exposure. The TRPV1-6 inhibitor
ruthenium red significantly reduced the Ca2+ response to
seawater challenge but did not abolish it (Fig. 3A). However, a potent selective
TRPV4 antagonist, HC067047, had no observed effect on the
seawater-induced increase of [Ca2+]c (Fig. 3B). Notably, the elevated level of
the [Ca2+]c response to seawater exposure was reduced by
treatment with the TRPV1-specific inhibitor, capsazepine (Fig. 3C). These results suggested that,
unlike ventilator-induced lung injury, extracellular
Ca2+ influx through TRPV1 channels was crucial to the
increase of [Ca2+]c observed in A549 cells following
exposure to seawater.
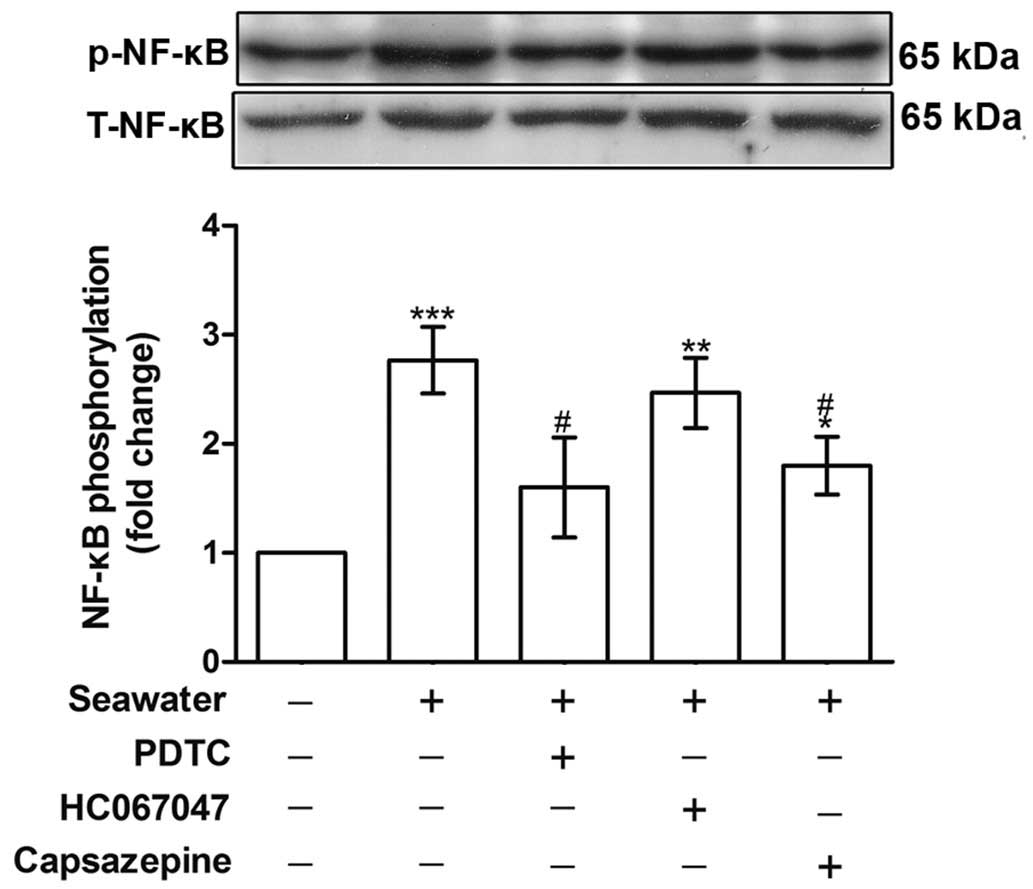
Seawater exposure induced TNF-α and IL-1β
release through TRPV1 activation and NF-κB phosphorylation
To establish the role of TRPV1 in mediating
Ca2+ influx and the subsequent inflammatory reactions,
the phosphorylation of NF-κB and the concentration of TNF-α and
IL-1β in the supernatant were measured. Fig. 4 indicates that NF-κB
phosphorylation was increased following seawater stimulation
(P<0.001, compared with the control group), whereas capsazepine
abolished this effect (P<0.05, compared with the seawater
treatment).
To understand how seawater stimulation induces
proinflammatory cytokine production, the effects of NF-κB and TRPV1
inhibition were compared. As presented in Fig. 5, cells pretreated with either PDTC
(NF-κB inhibitor) or, capsazepine (TRPV1 inhibitor) attenuated the
release of TNF-α and IL-1β elicited by the seawater challenge. PDTC
and capsazepine inhibited the increase in TNF-α and IL-1β
concentrations observed following seawater challenge. By contrast,
blockage of TRPV4 channels using HC067047 exhibited no effect on
the levels of these cytokines (Fig.
5). These results were consistent with the finding that
seawater challenge evoked extracellular Ca2+ influx by
activating TRPV1 channels rather than TRPV4.
Changes in the actin cytoskeleton of A549
cells exposed to seawater were diminished by calcium chelation
To explore the effect of seawater exposure on the
actin cytoskeleton, cells were fixed and stained to visualize the
actin cytoskeleton using Acti-stain 488 phalloidin. In the control
cells, actin filaments were observed to be in a regular arrangement
and evenly distributed in the cytoplasm (Fig. 6). By contrast, following seawater
challenge, cells exhibited a marked disorganization of actin
filaments, formation of stress fibers under the plasma membrane and
a dense ring of F-actin was located at the periphery of the cells
(Fig. 6). It has previously been
reported that cytosolic free Ca2+ oscillation can act as
a mediator of actin reorganization (24). To verify whether intracellular
calcium oscillation is a prerequisite for the remodeling of F-actin
under these conditions, intracellular calcium was chelated by the
preincubation of cells with BAPTA-AM or capsazepine. BAPTA-AM
partially reversed the disruption of the actin cytoskeleton.
However, the TRPV1-specific inhibitor, capsazepine, had no effect
on the F-actin distribution (Fig.
6).
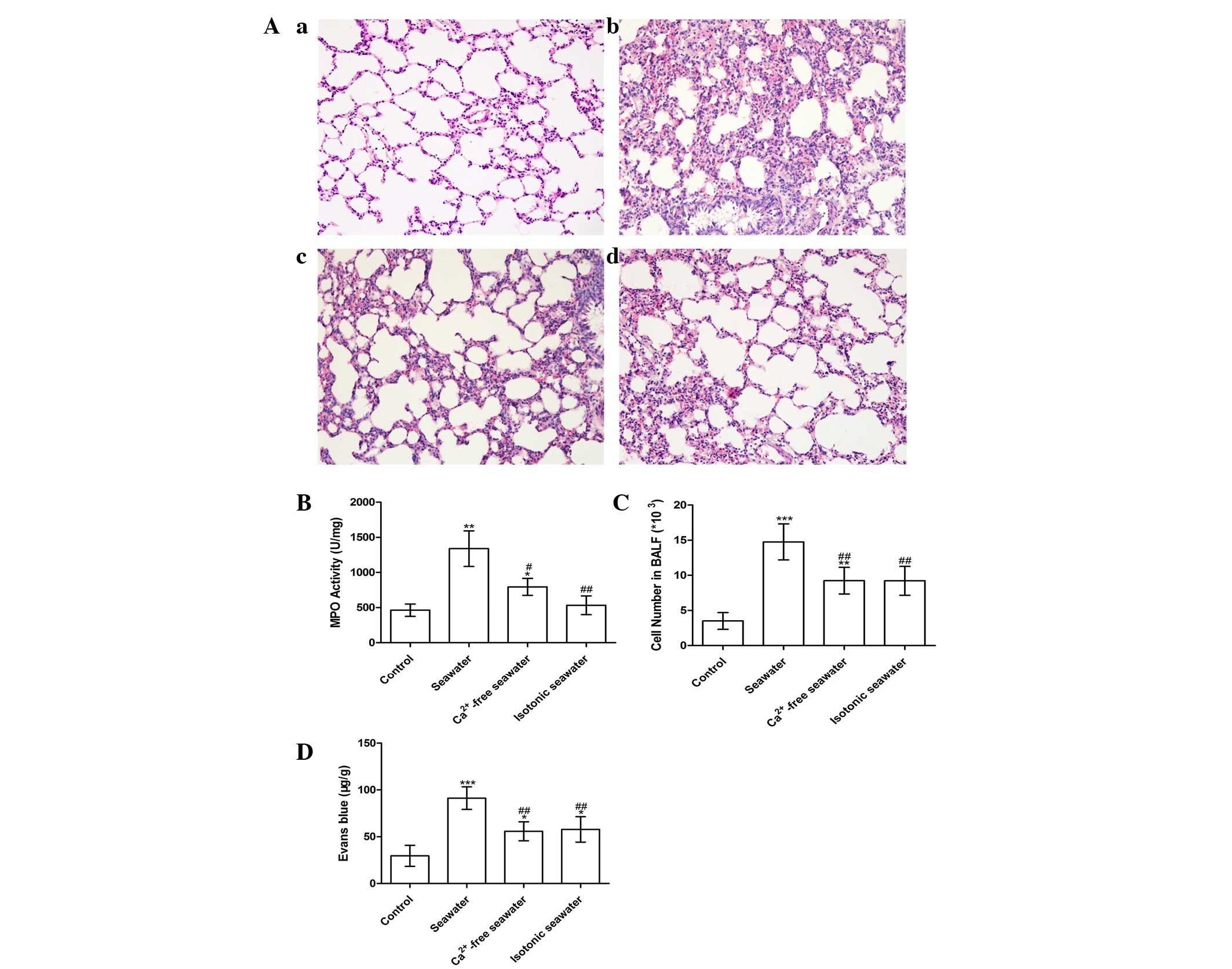
Seawater instillation induced lung injury
in a Ca2+-dependent manner
The pathology of seawater drowning-induced ALI is
characterized by simultaneous neutrophil infiltration, pulmonary
edema with hemorrhage, and production of inflammatory mediators
(25,26). Therefore, to assess the severity of
lung injury, TNF-α and IL-1β levels were examined, lung MPO
activity was measured to determine the levels of neutrophil
sequestration and histopathological examination of lung tissues was
conducted. These results are presented in Fig. 7. In order to investigate the effect
of high concentration of Ca2+ ions in seawater on the
severity of lung injury, the Ca2+ in seawater was
replaced with NaCl, followed by pH and osmotic pressure adjustment.
Histopathological examination of lung tissues exposed to seawater
displayed alveolar collapse, infiltration of inflammatory cells,
septal thickening and interstitial edema, demonstrating that
seawater challenge induced acute congestion in the lung tissues, in
addition to edema and inflammation (Fig. 7A). However, fewer changes to the
lung histoarchitecture were observed in the Ca2+-free
and isotonic seawater groups (Fig. 7Ac
and d).
The seawater group demonstrated significantly
increased MPO activity and number of the cells in the BALF, whereas
the Ca2+-free seawater and isotonic seawater groups
exhibited relatively lower levels compared with the seawater group
(Fig. 7B and C). To assess the
barrier permeability of the lung, the leak index of Evans blue dye
was assessed. The seawater instillation significantly increased the
barrier permeability. However, compared with the seawater group,
the Ca2+-free group and isotonic groups exhibited
significantly reduced barrier permeability (P<0.01; Fig. 7D). Consistent with these changes,
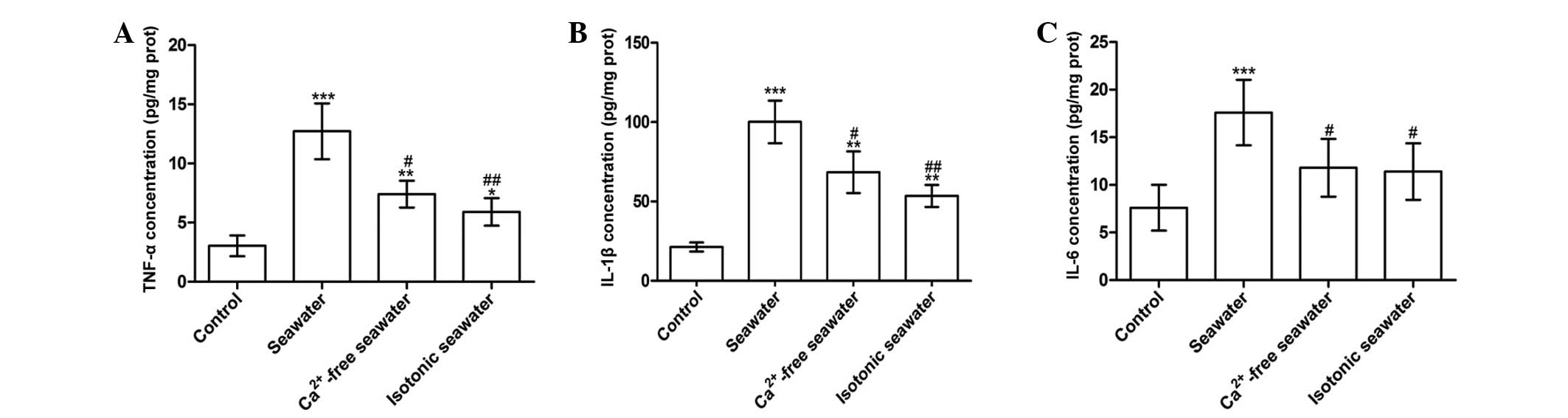
seawater instillation resulted in a significant increase of TNF-α,
IL-1β and IL-6 concentrations in lung tissues compared with control
rats (P<0.001; Fig. 8).
Compared with the seawater group, the levels of TNF-α and IL-1β
were reduced by instilling either Ca2+-free seawater or
isotonic seawater with no change in Ca2+ concentration
(Fig. 8).
TRPV1-mediated calcium oscillations
connect hypertonia signals and alterations during seawater
inhalation-induced lung injury
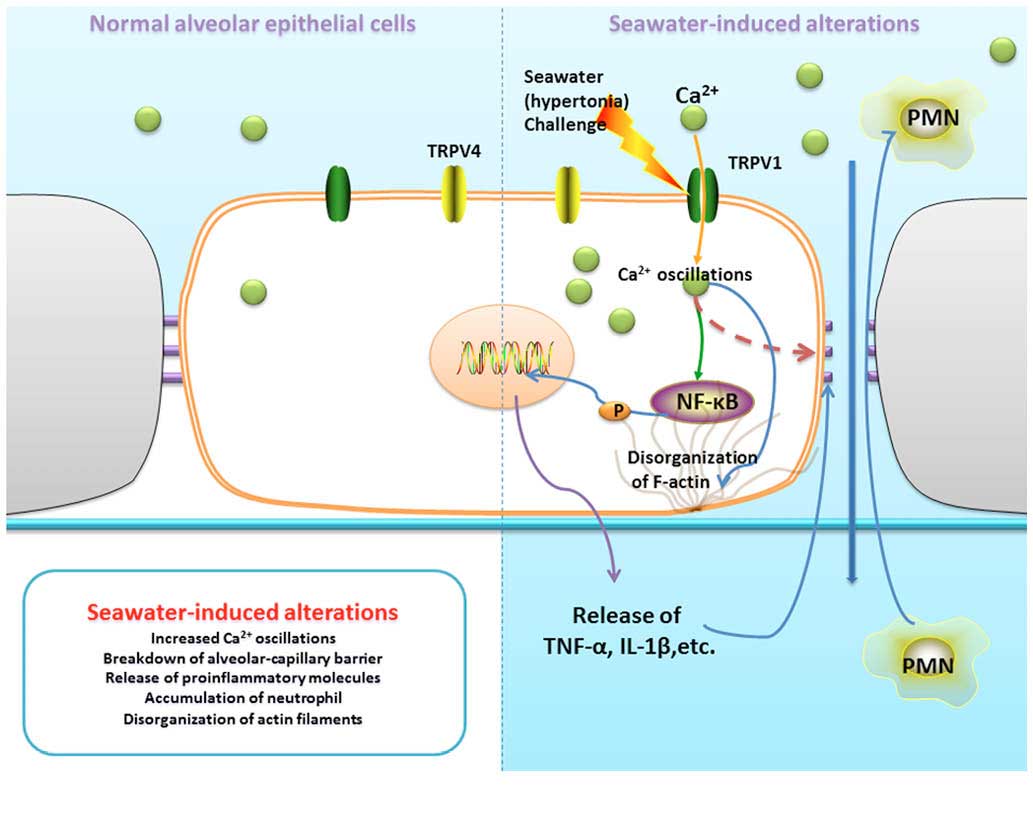
As summarized in Fig.
9, seawater challenge significantly increased Ca2+
oscillations in lung epithelial cells through the activation of
TRPV1. Furthermore, increased [Ca2+]c gives rise to the
breakdown of the alveolar-capillary barrier, release of
proinflammatory molecules, accumulation of neutrophils and
disorganization of actin filaments.
Discussion
Seawater drowning and associated ALI or respiratory
failure remain a notable cause of accidental death (25,26).
However, the underlying mechanism remains unclear, and requires
further exploration. Similar to lipopolysaccharide (LPS) and
cecalligation and puncture-induced lung injury, seawater
instillation can also induce excessive release of inflammatory
mediators, disturb the integrity of the alveolar septal network and
increase blood-air barrier permeability (4). However, seawater exposure directly
induces serious pulmonary interstitial edema, alveolar collapse,
disturbance of lung blood-air barrier permeability and subsequent
infiltration of inflammatory cells, in addition to the activation
of the pulmonary inflammatory cascade.
In the current study, the effect of calcium, a
component of seawater, on the inflammatory reactions in seawater
drowning-induced ALI was investigated. It was demonstrated that
high-concentration Ca2+ in seawater exacerbated lung
injury. Further study revealed that seawater challenge elevated
[Ca2+]c by inducing calcium entry from the extracellular
medium via TRPV1 channels. Elevated [Ca2+]c may have
induced the increased release of TNF-α and IL-1β. It was speculated
that these inflammatory reactions were associated with the
activation of NF-κB. Indeed, the study observed that the elevated
[Ca2+]c led to greater phosphorylation of NF-κB
(Fig. 4) and increased TNF-α and
IL-1β levels. This was corroborated by the diminished inflammatory
response following Ca2+ chelation, suggesting an
important role for cytosolic Ca2+ in the regulation of
lung inflammation.
Calcium is an important second messenger and
regulates a variety of cellular functions (6). It is recognized that ALI is often
dependent upon cytosolic free Ca2+ oscillation, and that
Ca2+ entry into lung endothelium can participate in
mediating microvascular-barrier permeability and the inflammatory
response to high vascular pressure, hydrogen sulfide or LPS.
Alvarez et al (6) reported
that disruption of the alveolar septal barrier resulting from
Ca2+ influx led to alveolar flooding and impaired gas
exchange. Consistent with these findings, Jian et al
(8) reported the HiPv-induced
increases in Kf were attenuated by low extracellular
Ca2+.
In the present experimental model, seawater
challenge resulted in a [Ca2+]c influx characterized by
rapid increase to a maximum level within 30 sec, followed by a
recovery period and sustained plateau. The source of
Ca2+ was clarified by chelation of Ca2+ in
the extracellular medium using EGTA, which resulted in a weakened
increase in [Ca2+]c following exposure to seawater, and
the result was confirmed by the re-addition of Ca2+ to
the cells. These results indicated that elevation of
[Ca2+]c evoked by seawater exposure was mainly
accomplished by increase of Ca2+ entry.
Various membrane transport pathways have been
identified as mediators of Ca2+ influx during ALI.
Tauseef et al (7)
demonstrated that endotoxins induce Ca2+ entry in
endothelial cells through the activation of transient receptor
potential canonical 6 channels in a Toll-like receptor 4-dependent
manner. Alvarez et al (6)
implicated TRPV4 in the Ca2+ entry-dependent regulation
of endothelial permeability, and the permeability response to the
TRPV4 agonist was abolished in lungs from TRPV4−/− mice.
TRPV1 was also reported to participate in sepsis-evoked ALI
(27). Pretreatment with
capsazepine markedly attenuated pulmonary COX-2 expression in
septic mice (27). To clarify
which channels were predominantly responsible for mediating
Ca2+ entry and the seawater-induced proinflammatory
cytokine production in A549 cells, the present study focused on the
role of TRPVs and blocked several potential pathways with the
inhibitors ruthenium red, capsazepine and HC067047 (Fig. 3). The results revealed that
extracellular Ca2+ influx required the activation of
TRPV1 channels following seawater challenge and may be
significantly reduced by the TRPV1-specific inhibitor, capsazepine,
and the TRPV family inhibitor, ruthenium red.
TRPV1 is a cell membrane-bound Ca2+
channel highly expressed in primary sensory neurons (28) and numerous other cell types,
including muscle cells, dendrites and airway epithelial cells
(12,19,28).
When cells are exposed to cytokines, abnormal pH, osmolality and
other irritations, intracellular calcium oscillates by activating
TRPV1 (12,18–20)
and can initiate endoplasmic reticulum stress and cell death in
human bronchial epithelial and alveolar cells (19). In cultured human lung cells, the
activation of TRPV1 by various stimuli can also promote
calcium-dependent cytokine release and acute respiratory
inflammation, with similar results reported in human corneal
epithelial cells (29).
Additionally, other studies have demonstrated that hypertonic
stress increased the levels of IL-6 and the chemoattractant IL-8 by
eliciting NF-κB activation in a TRPV1-dependent manner (29), and that TRPV1 activation altered
F-actin organization through extracellular regulated MAP kinase
(ERK1/2) and myosin light chain 2 (MLC2) pathways (30).
In view of the pivotal role of TRPV1 and
Ca2+ mobilization in the mediation of inflammation,
endoplasmic reticulum stress, cell death and reorganization of the
cytoskeleton, TRPV1 was selectively inhibited by capsazepine in
vitro to elucidate the function of TRPV1 on seawater
drowning-induced ALI. The results demonstrated that seawater
exposure gave rise to NF-κB phosphorylation and capsazepine or
Ca2+ chelation reduced the effect. Cells pretreated with
either capsazepine or an NF-κB inhibitor, PDTC, attenuated the
increase of TNF-α and IL-1β release elicited by seawater challenge.
Thus, seawater challenge may increase the release of
proinflammatory cytokines through the phosphorylation and
activation of NF-κB. A549 cells exhibited a marked disorganization
of actin filaments and formation of stress fibers following
exposure to seawater, whereas changes to the actin cytoskeleton
were diminished by pre-incubation of cells with the Ca2+
chelator, BAPTA-AM. However, pretreatment with the TRPV1-specific
inhibitor, capsazepine, produced no observed effect on the F-actin
distribution. It was surmised that this may be due to capsazepine
only being able to diminish calcium influx and ERK1/2 and MLC2
activation, rather than abolish them completely, a slight change to
Ca2+ influx may be sufficient to cause F-actin
redistribution. Furthermore, seawater may initiate such changes
through various other pathways. Further work is necessary to
elucidate this mechanism.
In conclusion, these observations place cytosolic
Ca2+ ions and TRPV1 at the center of the signaling
pathways that mediate seawater drowning-induced ALI, due to their
roles in modulating lung inflammation and the cytoskeleton. The
present study observed that high-concentration Ca2+ in
seawater exacerbated lung injury, and seawater challenge elevated
[Ca2+]c by activating TRPV1 channels, potentially
leading to the phosphorylation of NF-κB and subsequent increased
release of TNF-α and IL-1β.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant no. 81270124) and the
Military Key Projects in the 12th Five-year Plan of China (project
no. CWS13J043).
References
|
1
|
van Beeck EF, Branche CM, Szpilman D,
Modell JH and Bierens JJ: A new definition of drowning: Towards
documentation and prevention of a global public health problem.
Bull World Health Organ. 83:853–856. 2005.PubMed/NCBI
|
|
2
|
Engel SC: Drowning episodes: Prevention
and resuscitation tips. J Fam Pract. 64:E1–E6. 2015.PubMed/NCBI
|
|
3
|
Salomez F and Vincent JL: Drowning: A
review of epidemiology, pathophysiology, treatment and prevention.
Resuscitation. 63:261–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma L, Li Y, Zhao Y, Wang Q, Nan Y, Mu D,
Li W, Sun R, Jin F and Liu X: 3,5,4′-tri-O-acetylresveratrol
ameliorates seawater exposure-induced lung injury by upregulating
connexin 43 expression in lung. Mediators Inflamm. 2013:1821322013.
View Article : Google Scholar
|
|
5
|
Li J, Xu M, Fan Q, Xie X, Zhang Y, Mu D,
Zhao P, Zhang B, Cao F, Wang Y, et al: Tanshinone IIA ameliorates
seawater exposure-induced lung injury by inhibiting aquaporins
(AQP) 1 and AQP5 expression in lung. Respir Physiol Neurobiol.
176:39–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alvarez DF, King JA, Weber D, Addison E,
Liedtke W and Townsley MI: Transient receptor potential vanilloid
4-mediated disruption of the alveolar septal barrier: A novel
mechanism of acute lung injury. Circ Res. 99:988–995. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tauseef M, Knezevic N, Chava KR, Smith M,
Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE,
Dietrich A, et al: TLR4 activation of TRPC6-dependent calcium
signaling mediates endotoxin-induced lung vascular permeability and
inflammation. J Exp Med. 209:1953–1968. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jian MY, King JA, Al-Mehdi AB, Liedtke W
and Townsley MI: High vascular pressure-induced lung injury
requires P450 epoxygenase-dependent activation of TRPV4. Am J
Respir Cell Mol Biol. 38:386–392. 2008. View Article : Google Scholar
|
|
9
|
Pan Z, Wang Z, Yang H, Zhang F and Reinach
PS: TRPV1 activation is required for hypertonicity-stimulated
inflammatory cytokine release in human corneal epithelial cells.
Invest Ophthalmol Vis Sci. 52:485–493. 2011. View Article : Google Scholar :
|
|
10
|
Caterina MJ, Schumacher MA, Tominaga M,
Rosen TA, Levine JD and Julius D: The capsaicin receptor: a
heat-activated ion channel in the pain pathway. Nature.
389:816–824. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nishihara E, Hiyama TY and Noda M:
Osmosensitivity of transient receptor potential vanilloid 1 is
synergistically enhanced by distinct activating stimuli such as
temperature and protons. PLoS One. 6:e222462011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Geppetti P, Materazzi S and Nicoletti P:
The transient receptor potential vanilloid 1: Role in airway
inflammation and disease. Eur J Pharmacol. 533:207–214. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sadofsky LR, Ramachandran R, Crow C, Cowen
M, Compton SJ and Morice AH: Inflammatory stimuli up-regulate
transient receptor potential vanilloid-1 expression in human
bronchial fibroblasts. Exp Lung Res. 38:75–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thomas KC, Robers JK, Deering-Rice CE,
Romero EG, Dull RO, Lee J, Yost GS and Reilly CA: Contributions of
TRPV1, endovanilloids, and endoplasmic reticulum stress in lung
cell death in vitro and lung injury. Am J Physiol Lung Cell Mol
Physiol. 302:L111–L119. 2012. View Article : Google Scholar :
|
|
15
|
Hamanaka K, Jian MY, Weber DS, Alvarez DF,
Townsley MI, Al-Mehdi AB, King JA, Liedtke W and Parker JC: TRPV4
initiates the acute calcium-dependent permeability increase during
ventilator-induced lung injury in isolated mouse lungs. Am J
Physiol Lung Cell Mol Physiol. 293:L923–L932. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Chen L, Liedtke W and Simon SA:
Changes in osmolality sensitize the response to capsaicin in
trigeminal sensory neurons. J Neurophysiol. 97:2001–2015. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sidhaye VK, Guler AD, Schweitzer KS,
D'Alessio F, Caterina MJ and King LS: Transient receptor potential
vanilloid 4 regulates aquaporin-5 abundance under hypotonic
conditions. Proc Natl Acad Sci USA. 103:4747–4752. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johansen ME, Reilly CA and Yost GS: TRPV1
antagonists elevate cell surface populations of receptor protein
and exacerbate TRPV1-mediated toxicities in human lung epithelial
cells. Toxicol Sci. 89:278–286. 2006. View Article : Google Scholar
|
|
19
|
Thomas KC, Sabnis AS, Johansen ME, Lanza
DL, Moos PJ, Yost GS and Reilly CA: Transient receptor potential
vanilloid 1 agonists cause endoplasmic reticulum stress and cell
death in human lung cells. J Pharmacol Exp Ther. 321:830–838. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee LY and Gu Q: Role of TRPV1 in
inflammation-induced airway hypersensitivity. Curr Opin Pharmacol.
9:243–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guler AD, Lee H, Iida T, Shimizu I,
Tominaga M and Caterina M: Heat-evoked activation of the ion
channel, TRPV4. J Neurosci. 22:6408–6414. 2002.PubMed/NCBI
|
|
22
|
Liedtke W, Choe Y, Marti-Renom MA, Bell
AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM and Heller S:
Vanilloid receptor-related osmotically activated channel (VR-OAC),
a candidate vertebrate osmoreceptor. Cell. 103:525–535. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O'Neil RG and Heller S: The
mechanosensitive nature of TRPV channels. Pflugers Arch.
451:193–203. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rosado JA, González A, Salido GM and
Pariente JA: Effects of reactive oxygen species on actin filament
polymerisation and amylase secretion in mouse pancreatic acinar
cells. Cell Signal. 14:547–556. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma L, Zhao Y, Li B, Wang Q, Liu X, Chen X,
Nan Y, Liang L, Chang R, Liang L, et al:
3,5,4′-Tri-O-acetylresveratrol attenuates seawater
aspiration-induced lung injury by inhibiting activation of nuclear
factor-kappa B and hypoxia-inducible factor-1α. Respir Physiol
Neurobiol. 185:608–614. 2013. View Article : Google Scholar
|
|
26
|
Fan Q, Zhao P, Li J, Xie X, Xu M, Zhang Y,
Mu D, Li W, et al: 17β-Estradiol administration attenuates seawater
aspiration-induced acute lung injury in rats. Pulm Pharmacol The.
24:673–681. 2011. View Article : Google Scholar
|
|
27
|
Ang SF, Sio SW, Moochhala SM, MacAry PA
and Bhatia M: Hydrogen sulfide upregulates cyclooxygenase-2 and
prostaglandin E metabolite in sepsis-evoked acute lung injury via
transient receptor potential vanilloid type 1 channel activation. J
Immunol. 187:4778–4787. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cortright DN and Szallasi A: Biochemical
pharmacology of the vanilloid receptor TRPV1. An update. Eur J
Biochem. 271:1814–1819. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reilly CA, Taylor JL, Lanza DL, Carr BA,
Crouch DJ and Yost GS: Capsaicinoids cause inflammation and
epithelial cell death through activation ofvanilloid receptors.
Toxicol Sci. 73:170–181. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cong X, Zhang Y, Yang NY, Li J, Ding C,
Ding QW, Su QC, Mei M, Guo XH, Wu LL and Yu GY: Occludin is
required for TRPV1-modulated paracellular permeability in the
submandibular gland. J Cell Sci. 26:1109–1121. 2013. View Article : Google Scholar
|